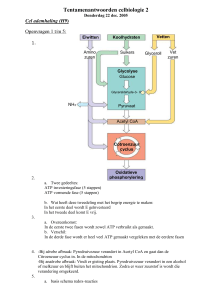
Mitochondrion - WordPress.com
advertisement

MITOCHONDRION http://home.kpn.nl/b1beukema/mitochondrien.html Mitochondrion/driën Van: TJEERDO (Oorspronkelijk bericht)17/01/2005 1 Oorsprong 2 Mitochondriaal DNA 3 Terug in de tijd met Mitochondriën 4 Nadelen 5 Externe link http://www.ronaldschulte.nl/Preparaten%20De%20cel%2002.htm http://www.ronaldschulte.nl/Preparaten%20De%20cel%2003.htm Afbeelding Mitochondrion 1. Inwendig membraan. 2. Uitwendig membraan. 3. Crista. 4. Matrix. Dit is een mitochondrion. De buitenste membraan valt onder eukaryotische origine. De binnenste membraan is volgens de endosymbiont-theorie van prokaryotische oorsprong. Dit was een prokaryotische bacterie die door fagocytose opgenomen is in de eukaryotische cel. Het binnenste membraan is de locatie waar het elektronentransportsysteem zich bevindt. De geplooide delen van het membraan wordnen cristae genoemd. De ruimte tussen de membranen is de plek waar de mitochondriele ademhalingsprocessen plaatsvinden. De ademhalingsketen is een transportketen voor elektronen. De energie die hieruit voortvloeid, kan gebruikt worden in het proces wat oxidatieve fosforylatie wordt genoemd. Met als einddoel ATP-synthese. Het proces is ook bekend onder de naam citroenzuurcyclus of Kreb's -cyclus ATP Acetyl-CoA /co -enzyme A /citroenzuurcyclus Het fascinerende aan het mitochondriele erfelijke systeem is dat het typisch bacteriele eigenschappen heeft. Het DNA is cirkelvormig (70S) evenals bacterie-DNA. De ribosomen zijn kleiner dan de cytoplasma-ribosomen en hebben ongeveer hetzelfde formaat als bacteriele ribosomen. Hun vermogen om eiwitten te synthetiseren wordt geremd door het antibioticum chlooramfenicol, een stof die de eiwitsynthese van bacterien remt maar niet die van eukaryotische organismen. Verder worden mitochondrien niet aangetast door de eukaryotische remmer cycloheximide, die ook geen effect heeft op bacterien. De mitochondrien gebruiken een andere sequentieele code dan de rest van de cel. Mitochondrion http://nl.wikipedia.org/wiki/Mitochondrion Een mitochondrion of mitochondrium (meervoud mitochondriën of mitochondria) is een staaf- of bolvormig celorganel dat een dubbel membraan bevat en in het cytoplasma van de cel ligt. De vorm kan ook onregelmatig zijn. De grootte is meestal 1 micrometer. De mitochondriën zijn de energiecentrales van het lichaam. Het aantal mitochondriërn per cel staat dan ook in relatie tot de energiebehoefte van de cel. Deze energie komt beschikbaar in de vorm van de stof ATP, een gebruikelijke energierijke stof in de hele biochemie, die aangemaakt wordt bij het omzetten van suikers en vetzuren in water en CO2. Dit proces kan enkel plaats vinden in aanwezigheid van zuurstof en gebeurt in een proces dat citroenzuurcyclus (of Krebs cyclus) wordt genoemd. Het mitochondrion is de enige plaats in de cel waar oxidatie met zuurstof (aerobe stofwisseling) kan plaatsvinden. Een mitochondrium bezit een dubbel biologisch membraam: een uitwendig membraan een inwendig membraan, dat instulpingen, de cristae, vertoont. Tussen de cristae zit de matrix (vloeistof). Het aantal cristae staat ook in relatie met de intensiteit van de ademhaling. De vermenigvuldiging van de mitochondriën gebeurt door tweedeling. Mitochondriaal DNA De mitochondriën bevatten eigen genetisch materiaal in de vorm van een klein ringvormig DNA-molecule. De mitochondriën bevatten ook de ribosomen. De genetische eigenschappen in mitochondriaal DNA erven normaliter uitsluitend over via de vrouwelijke lijn (een eicel beval namelijk wel moederlijke mitochondria, maar een zaadcel geen vaderlijke), en geven zo bijzondere informatie over de afstamming. Enkele voorbeelden van overdracht van mitochondriaal DNA van de vader zijn echter wel beschreven, hoewel tot vrij recent werd aangenomen dat dit niet kon. Het gegeven van de moederlijke oorsprong van alle mitochondriaal DNA is gebruikt om te proberen de oorsprong van de oudste voorvader van de mens te traceren (of eigenlijk dus de oudste voormoeder). Deze hypothetische 'Eva' van wie alle mensen op de wereld uiteindelijk afstammen lijkt uit Afrika te zijn gekomen, ca 200.000 (?) jaar geleden. De datering hangt sterk af van de schattingen van de frequentie waarmee toevallige mutaties in het mitochondriaal DNA optreden. Oorsprong Er bestaat een aannemelijke hypothese, de endosymbiontenhypothese, dat mitochondria, net zoals chloroplasten bij planten, oorspronkelijk vrijlevende bacteriën waren die al in een vroeg stadium in de evolutie intracellulair gingen leven in symbiose met hun gastheer. Externe Link [1] (http://www.homepages.hetnet.nl/~b1beukema/mitochondrien.html) mitochondrion en de endosymbiose-theorie Mitochondriaal DNA http://nl.wikipedia.org/wiki/Mitochondriaal_DNA Mitochondriaal DNA of mtDNA is klein ringvormig DNA dat zich niet in de celkern bevindt, maar in de mitochondriën. De mitochondriën bevatten ook een eigen type ribosomen. Er zijn honderden mitochondri챘n per cel en deze bevatten elk vele circulaire chromosomen. Deze bevatten alle dezelfde genen, maar zijn niet steeds identiek: isodisomie wil zeggen dat alle moleculen van 1 cel identiek zijn, heterodisomie betekent dat ze niet-identiek zijn. Het dubbelstrengige mtDNA bij de mens bevat 37 genen: 22 coderen tot tRNA, 2 tot ribosomaal-RNA en de overige 13 tot eiwitten. Overerving Het mitochondriaal DNA erft normaliter uitsluitend over via de vrouwelijke lijn en gaat dus niet over met het sperma of stuifmeel. De mitochondriën van de mannelijke zaadcel bevinden zich in de staart, die bij de bevruchting meestal niet de eicel binnentreedt. Enkele voorbeelden van overdracht van mitochondriaal DNA van de vader zijn echter wel beschreven. Het gegeven van moederlijke (maternale) overerving van mtDNA wordt gebruikt in de genografie om de verspreiding van onze voorouders (of eigenlijk de voormoeders) in de prehistorie in kaart te brengen. Deze hypothetische 'Eva' van wie alle mensen op de wereld uiteindelijk afstammen lijkt uit Afrika te zijn gekomen, ca 140.000 tot 200.000 jaar geleden. De datering hangt sterk af van de schattingen van de frequentie waarmee toevallige mutaties in het mitochondriaal DNA optreden. Bij het in kaart brengen van de migratie van de zomereik naar het Noorden na de laatste ijstijd is ook gebruikgemaakt van mtDNA. Aandoeningen Een mitochondriale overerving is kenmerkend voor bepaalde aandoeningen, omdat deze enkel langs de moeder kunnen worden doorgegeven: het kind krijgt al het cytoplasma van de moeder, en hierin bevinden zich de mitochondriën. Deze aandoeningen komen vaak voor in lichaamsdelen en organen die veel energie nodig hebben (hersenen, hart, ogen, oren,...), daar de mtDNA voor de oxidatieve fosforylering zorgt (energievoorziening in de cel). Mitochondriën vertellen over de evolutie http://www.kennislink.nl/web/show?id=16361&vensterid=811&cat=60274 SAMENVATTING Wat gebeurde er anderhalf miljard jaar geleden? Canadese wetenschappers lazen het in DNA van een eencellig organisme. Deze recente ontdekking ontrafelt de basismechanismen van de evolutie. Een missing link toont hoe cellen evolueren. / Drs Ann De Ron/ Natuur & Techniek /01 september 1997 Op 29 mei 1997 meldt het tijdschrift Nature de ontdekking van een ‘oermitochondrion’. De groep rond vijf wetenschappers van drie afzonderlijke Canadese universiteiten vond een missing link tussen ver geëvolueerde mitochondriën en hun bacteriële voorouders. Het lijkt wel of de evolutie het mitochondrion in kwestie zo goed als onberoerd heeft gelaten. “Een kind dat de moleculaire speeltuin heeft gemist”, heet het in een commentaar in hetzelfde blad. Onze planeet, meer dan anderhalf miljard jaar geleden. In de atmosfeer neemt het zuurstofgehalte toe. Het leven bestaat uit cellen met een primitief ontwerp: prokaryoten. Toevallig dringt een van deze cellen binnen in een andere. De twee cellen beginnen een symbiotische levenswijze: ze leven samen en ondervinden daar beide voordeel van. Daarmee is de eukaryote cel geboren. De opgenomen cel, de endosymbiont, bevat naar schatting 500 genen maar verliest het grootste deel van deze stukjes DNA in de loop van de evolutie. Wat rest is het mitochondrion zoals we het nu kennen, de energiecentrale van de cel. Een cel eet een bacterie op. In een tweede fase blijft de bacterie behouden in een voedselblaasje van de cel. In fase drie is de bacterie geëvolueerd tot een mitochondrion. Zo goed als alle wetenschappers nemen deze endosymbiotische theorie ondertussen voor waar aan. De theorie verklaart het ontstaan van mitochondriën en het feit dat ze over eigen DNA beschikken. De bekende mitochondriën verschillen onderling sterk. De vorm van het oorspronkelijke mitochondrion kun je er niet uit afleiden. Daarvoor was het wachten op een mitochondrion waar de evolutie minder vat op heeft gehad. Driedimensionale weergave van een mitochondrion dat in de lengte is doorgesneden. Kenmerkend is de dubbele membraan, waarvan de binnenste instulpingen vertoont. In het centrale gedeelte ligt het mitochondriaal DNA. Reclinomonas americana Met de Canadese ontdekking is het zover. Het mitochondrion van Reclinomonas americana, een eencellig zoetwaterorganisme, lijkt sterk op het 500 genen tellende mitochondrion uit voorhistorische tijden. Reclinomonas-mitochondriën bevatten 97 genen, een ongeëve-naard record. Daarvan coderen er 62 voor een eiwit met een bekende functie. Menselijke mitochondriën bevatten er slechts dertien, die van gist acht en die van de malariaparasiet drie. Alle mitochondriale genen die eerder bekend waren, komen terug op het mitochondriaal DNA van Reclinomonas. Dat versterkt de veronderstelling dat mitochondriën niet meer dan één keer in de evolutiegeschiedenis ontstonden. Een aantal van de Reclinomonas-genen is nooit eerder in mitochondriaal DNA teruggevonden. Michael Gray, de onderzoekscoördinator van het Canadese team: “Onze ontdekking kan wel eens de laatste restjes verzet tegen de endosymbiotische theorie verjagen.” Alles wijst erop dat het Reclinomonas-mitochondrion een missing link is tussen bacteriën en moderne mitochondriën Weergave van het eencellige waterorganisme Reclinomonas americana. Fossielen “Theorieën over evolutie zijn altijd een beetje onaangenaam”, reageert de Gentse hoogleraar genetica en biotechnologie Marc Van Montagu op de Canadese ontdekking. “Ze vallen niet te bewijzen. Daarom is het vinden van een tussenschakel zo belangrijk.” Een fossiel kan zo’n tussenschakel zijn. Van de eerste vogel, Archaeopterix, werd eerst een enkel veertje en later het volledige fossiel teruggevonden. Het uitgestorven dier bevindt zich op de overgang tussen reptielen en vogels. Levende fossielen bestaan ook. De coelacanth-vis zwemt nog rond en heeft ‘kwastvinnen’ die een voorloper zijn van poten. Het is een overgangsdier tussen primitieve vissen en de hogere gewervelde dieren. Evolutiestamboom Zowel Archaeopterix als de coelacanth zijn belangrijk om een beeld te krijgen van hoe de evolutie te werk ging. Het zijn links tussen bestaande groepen, vertakkingspunten op een evolutieboom. Een evolutieboom geeft de evolutionaire verbanden tussen organismen weer, zoals een familiestamboom dat doet voor de leden van een familie. Helemaal onderaan de evolutieboom zijn er waarschijnlijk drie aftakkingen. De ene tak leidt naar moderne Eubacteriën, de tweede naar Archaebacteriën en de derde naar organismen met eukaryote cellen. Weten hoe de eerste primitieve eukaryote cel eruit zag, is al even moeilijk als zich een voorstelling maken van het eerste mitochondrion. Het een kan trouwens niet zonder het ander. Intermezzo De cel Levende organismen zijn onder te verdelen in prokaryoten en eukaryoten. Bacteri챘n zijn prokaryoten. Binnen eukaryoten vallen vier groepen organismen te onderscheiden: dieren, planten, schimmels en protoctisten. Protoctisten zijn domweg alle eukaryoten die geen plant of dier of schimmel zijn. De eerste cellen op aarde leken op de huidige prokaryote cellen. Prokaryote cellen zijn eenvoudiger van structuur en kleiner dan eukaryote cellen. Een prokaryote cel is van de buitenwereld gescheiden door een dun vlies, de celmembraan, maar heeft geen inwendige membranen. Een eukaryote cel heeft wel een membraan rond de kern en rond een aantal celstructuren, de organellen. Deze organellen vervullen belangrijke functies voor de cel. Het bladgroen van planten zit in organellen die instaan voor de fotosynthese: choroplasten. Minstens even belangrijk zijn mitochondriën, organellen die in zo goed als elke eukaryote cel voorkomen. Leven op zuurstof is mogelijk dankzij mitochondriën. Het zijn de ‘batterijtjes’ van de cel; ze zorgen voor de productie van energie. Zowel chloroplasten als mitochondriën bevatten eigen DNA. Eukaryote cel van een zoogdier. Evolutieboom naar de inzichten van Gertraud Burger. Aantal en lengte van vertakkingen hebben geen betekenis. Einde Intermezzo DNA In de kern zit erfelijk materiaal van de cel. Mitochondriën zijn weliswaar onderdelen van een cel maar bevatten eigen DNA. Dat bleek zo’n 35 jaar geleden al. DNA is een dubbele keten van fosfaten en suikers waartussen basenparen hangen. In 1992 was de basenvolgorde (sequentie) van het mitochondriaal DNA nog maar van een beperkt aantal organismen volledig bekend. Het ging om enkele dieren, een paar schimmels en één plant. Een aantal Canadese onderzoekers richtte in 1992 het Organel Genoom Megasequencing Programma (OGMP) op. Opzet: de volledige basenvolgorde ophelderen van het mitochondriaal DNA van een brede selectie eencellige eukaryoten, de protoctisten. Het OGMP-team stelde zich een dubbel doel. Langs de ene kant wilde het meer informatie verzamelen over de ontstaansrelaties tussen die protoctisten. Dat draagt bij tot het opstellen van een globale evolutieboom van het leven. Parallel daaraan ging de interesse uit naar de evolutie van mitochondriën zelf. Het spreekt vanzelf dat het onbegonnen werk is de mitochondriën van alle organismen in detail te bekijken. Het gaat erom een goede keuze te maken. Reclinomonas americana is een voltreffer. Het is een eencellig zoetwaterorganisme dat zich voedt met bacteriën. De ‘primitieve aanblik’ ervan wekte de belangstelling van dr Charles O’Kelly, verbonden aan een Amerikaans laboratorium voor oceaanonderzoek. O’Kelly vertelt hoe hij Reclinomonas koos: “Eigenlijk vond het mij. In 1989 kreeg ik besmetting in een cultuur. Om te vinden wat mij daar stoorde, bekeek ik het onder de microscoop.” Hij beval het OGMP-team het interessante organisme aan. Analyse Het uitverkoren organisme arriveert op het laboratorium. Een moleculair-bioloog bepaalt de basenvolgorde van het mitochondriaal DNA. Daartoe isoleert hij een groot aantal mitochondriale DNA moleculen. Vervolgens breekt hij die moleculen in stukken. Dat gebeurt willekeurig; elk molecuul breekt op andere plaatsen. Van elk stuk bepaalt hij de basenvolgorde. Zo verkrijgt de onderzoeker een aantal flarden van de volledige basenvolgorde. Doordat de moleculen telkens anders breken, overlappen de basenvolgorden gedeeltelijk. Een computer springt bij om de flarden te ordenen en aan elkaar te plakken. Zo komt men tot de basenvolgorde van het volledige molecuul. Dan volgt de statistische verwerking. Bekende mitochondriale basenvolgorden van Reclinomonas en andere organismen worden op alle mogelijke manieren met elkaar vergeleken, in de hoop betrouwbare conclusies te trekken over evolutionaire verbanden. Ellenlange rijen letters in de trant van ATTACCGCAT; wie doet er iets mee? De Amsterdamse hoogleraar Les Grivell kan zich goed voorstellen dat hij de sequentie ooit raadpleegt. “We gebruiken vaak aanwijzingen uit de evolutie om iets meer te weten te komen over de functie van een eiwit.” Zijn vakgroep Moleculaire Celbiologie van de Universiteit van Amsterdam kijkt hoe het mitochondriaal DNA van bakkersgist tot expressie komt en vooral hoe de cel het “op een heel nette manier voor elkaar krijgt” om haar twee erfelijke systemen te laten samenwerken. Vervolg vragen Dr Yves Van de Peer van de universiteit Antwerpen (UIA) vindt het Nature-artikel vooral interessant in het kader van de evolutie van organellen. Hij ziet in het artikel “weinig nieuws” wat betreft bevestiging van de endosymbiotische theorie; daar waren al eerder genetische aanwijzingen voor. “Al in 1985 verscheen een sleutelpublicatie en ook onze onderzoeksgroep publiceerde in 1995 een artikel waarin we de bacteriële oorsprong van mitochondriën nogmaals aantonen.” Grivell reageert: “Het artikel is wel degelijk heel nieuw. Tot nu toe had je altijd het idee dat er een grote kloof is tussen bacteriën en wat je nu vindt bij mitochondriën. Dit mitochondrion is bevroren in de tijd, als een fossiel in amber.” Over de betekenis van de Canadese ondekking vervolgt Grivell: “Het artikel levert verrassende inzichten over mitochondriale evolutie. Het geeft voor een deel antwoorden maar maakt het ook gemakkelijk om nieuwe vragen te formuleren. In de natuur kan veel meer gebeuren dan in een lab. De natuur heeft erfelijkheidsexperimenten in onze plaats gedaan. Tot nu toe waren protoctisten een achtergebleven gebied in onderzoek.” Volgens Grivell kan het protoctistenonderzoek wel eens in een stroomversnelling terechtkomen. Het OGMP-team zelf, hoopt verder te werken aan de evolutieboom van protoctisten. Er blijven nog veel vragen onbeantwoord. Zo is het nog onzeker hoe vroeg in de eukaryote evolutie mitochondriën ontstonden. Gertraud Burger, hoofd van het centraal laboratorium van OGMP: “We hebben nu al een aantal soorten aan de uiteinden van de boom onderzocht. Je zou kunnen spreken van de ‘bladeren’ van de evolutieboom. Nu gaan we ons concentreren op de ‘wortel’, door te kijken naar de mitochondriën van heel primitieve organismen.” Het team wil ook starten met de studie van bacteriën om later het volledige genoom te bepalen van de kandidaat die het meest verwant is aan mitochondriën. “We komen van twee kanten, van onder de grond tot aan de wortel en van de bladeren tot aan de wortel”, lacht Burger. Consumptie Burger laat zich verleiden tot een speculatie over de verre toekomst. “Als we dichter naderen tot het moment waarop een bacterie in een andere cel ging leven, kunnen we beter begrijpen hoe symbiose werkt. De vraag hoe aparte organismen tezamen komen en zo’n intieme relatie vormen is uiterst intrigerend. Dát begrijpen geeft ook inzicht in manieren om symbiose te manipuleren. Misschien wil je twee organismen combineren die totaal andere dingen doen, op een manier dat ze samen functioneren.” Soms staan honderden genen samen in voor een enkel kenmerk. De huidige biotechnologie kan een enkel gen inbrengen, maar is in zo’n geval voorlopig machteloos. Burger vervolgt: “Voor een complex kenmerk zou ‘kunstmatige symbiose’ de te volgen weg zijn. Zo zou je in gist een geschikte bacterie kunnen binnenbrengen om alcohol te produceren uit zonlicht in plaats van uit suiker. Ik zie de toepassing van die alcohol dan wel als brandstof en niet voor consumptie. Maar neem dit voorbeeld niet te serieus”, waarschuwt ze. Toepassingen van een ontdekking voorspellen, is misschien nog moeilijker dan ermee in het verleden kijken. Intermezzo DNA uit kern of mitochondrion? De OGMP-wetenschappers bepalen basenvolgorden van mitochondriaal DNA. Dat is logisch vanuit hun interesse voor de evolutie van mitochondriën. Ze gebruiken hetzelfde materiaal ook om evolutionaire relaties tussen de organismen zelf op te helderen. Toch is daarvoor de analyse van mitochondriaal DNA alléén, niet zaligmakend. Gertraud Burger van OGMP: “Een eukaryoot organisme bevat minstens twee genomen: het kerngenoom en het mitochondriaal genoom. Elk van die twee vertegenwoordigt tot op zekere hoogte de evolutiegeschiedenis van het volledige organisme. Alleen een combinatie van beide geeft ons een volledig beeld.” Burger verdedigt de keuze van OGMP voor mitochondriaal DNA als vergelijkingsmateriaal: “Mitochondriaal DNA is een perfect molecuul om evolutiebomen op te stellen, aangezien je daar een vergelijkbare set genen hebt die altijd op dezelfde plaats zitten. In het kerngenoom daarentegen is het vinden van vergelijkbare genen in verschillende organismen technisch moeilijk. Als alternatief kun je het volledige kerngenoom van een aantal organismen bepalen, maar dan praten we over een ontzaglijke hoeveelheid werk.” Toch vinden veel wetenschappers het kerngenoom beter geschikt om genetische stambomen te maken. Prof dr Piet Borst, wetenschappelijk directeur van het Nederlands Kanker Instituut en geïnteresseerd in de vorming van celorganellen, vindt vergelijkingsmateriaal uit de kern “op termijn betrouwbaarder”. Een veel gehoorde uitspraak is dat het kerngenoom eerder geschikt is om ‘lange afstanden’ te analyseren, terwijl de toepassingen van mitochondriaal DNA zich eerder op ‘kortere afstanden’ situeren. ‘Afstanden’ verwijst naar de mate waarin organismen door de evolutie van elkaar gescheiden zijn. Zo is de evolutionaire afstand tussen mensen en beukenbomen groot en die tussen mensen en apen relatief klein. Voorbeelden van het gebruik van mitochondriaal DNA op ‘korte afstanden’ zijn er genoeg. Door het gebruik van mitochondriaal DNA kreeg de speurtocht naar het ontstaan van Homo sapiens in 1987 een nieuwe impuls, en bleek dit jaar dat de Neanderthaler geen voorouder van ons is. Mitochondriaal DNA vertelde de onderzoeksgroep evolutionaire biologie van het Universitair Centrum Antwerpen (RUCA) over de manier waarop muizen zich over Europa hebben verspreid. Onderzoek over lange evolutie-afstanden, gebruikt voor het opstellen van evolutiebomen, baseert zich tot nu toe vooral op klein ribosomaal DNA uit de kern. Burger wijst op de grote hoeveelheid volledig bekende mitochondriale DNA’s. “We kunnen evolutiebomen samenstellen die vijfmaal meer gegevens per soort bevatten dan de huidige standaard evolutiebomen. Daarom konden ‘onze’ bomen controversiële vertakkingen ophelderen.” Dr Thierry Backeljau, verbonden aan het Koninklijk Belgisch Instituut voor Natuurwetenschappen, ziet het gebruik van volledige mitochondriale basenvolgorden als een recent, krachtig en veelbelovend middel. Een onderzoeker krijgt meer dan een kale basenvolgorde. Hij krijgt waardevolle informatie over volgorde en oriëntatie van een heel aantal genen van diverse typen, waarbij elk type met zijn eigen snelheid evolueert. “Dat verdient zeker de voorkeur boven het bekijken van een enkel gen uit de kern.” The origin and early evolution of mitochondria http://genomebiology.com/2001/2/6/REVIEWS/1018 2010 : Eukaryote & prokaryote cellen = mitochondrion de sleutel tot meercelligheid en groot , maar rommelig genoom http://scienceblogs.com/pharyngula/2010/12/how_to_afford_a_big_sloppy_gen.php Creationists ; Creationism Slips Into a Peer-Reviewed Journal http://ncseweb.org/rncse/28/3/creationism-slips-into-peer-reviewedjournal http://genefinding.blogspot.com/2008/02/stealth-attempt-to-sneak-creationism.html. Attila Cordas http://pimm.wordpress.com/2008/01/29/http://pimm.wordpress.com/2008/01/29/ can-you-tell-a-good-article-from-a-bad-article-based-on-the-abstract-and-the-title/ Lars Juhl Jensen http://larsjuhljensen.wordpress.com/2008/02/10/commentary-neither-buried-nor-treasure PZ Myers Pharyngula blog http://scienceblogs.com/pharyngula/2008/02/a_baffling_failure_of_peer_rev.php http://scienceblogs.com/pharyngula/2009/02/whatever_happened_with_the_pro.php wanneer-twee-genomen-elkaar-mislopen http://ascendenza.wordpress.com/2011/10/24/wanneer-twee-genomen-elkaar-mislopen/#comments Bijna alle levensvormen bezitten twee genomen. Wanneer deze niet goed op elkaar afgestemd zijn kunnen er nare gevolgen zijn zoals snelle veroudering, ziekten als Alzheimer en kanker. Deze twee genomen bestaan uit het nucleaire en het mitochondriale genoom. Het nucleaire genoom verwijst naar het DNA waar we het doorgaans over hebben als we spreken van 'de genen'. Maar een minstens zo belangrijk onderdeel van onze cellen zijn de mitochondriën. Ook zij bevatten een weliswaar klein genoom wat exact moet samenwerken met dat van de nucleus STRONG>Hoewel de oceanen op Aarde zuurstofatomen in hun watermoleculen bevatten, was er in eerste instantie nog geen spoor van moleculair zuurstof (O2) te bekennen. De eerste bacteriën waren dan ook verplicht anaeroob. Enkele van deze bacteriën ontwikkelden de mogelijkheid de energie van het zonlicht te gebruiken door middel van fotosynthese. Zij produceerden zuurstof dat voor de anaerobe bacteriën giftig was. Enkele van deze bacteriën die zuurstof schuwden evolueerden de mogelijkheid om deze zuurstof te ademen. Deze laatste bacteriën vormen de voorouders van de mitochondriën. Een aantal werd door cellen verzwolgen en dit revolutioneerde het leven op Aarde; de eerste eukaryoten ofwel de eerste meercelligen konden zodoende ontstaan.* Eukaryoot Mitochondriën zijn organellen in de cel die verantwoordelijk zijn voor de ademhaling van de cel en het organisme. Alle biologen zijn het er inmiddels over eens dat deze mitochondriën afstammen van endosymbiontische bacteriën. Ze bezitten net als de aerobe bacteriën een elektronentransportketen in hun membraan die uiteindelijk zuurstof reduceert tot water. Het mitochondrion heeft een eigen genoom. Dit genoom is gereduceerd vergeleken met dat van een bacterie en bevat de meest essentiële genen voor de interne functies van het mitochondrion. Het genoom van de mitochondriën is helemaal afgestemd op dat van de celkern. Beide genomen produceren de eiwitten die in de mitochondriën zorg dragen voor het elektronentransport. Deze eiwitten moeten perfect samenwerken. Is er een kleine discrepantie, veroorzaakt door een puntmutatie bijvoorbeeld, dan lekt het elektronentransport elektronen en worden er vrije radicalen gevormd. Mitochondriën worden via de moeder doorgegeven in plaats van door beide ouders zoals het geval is met het nucleaire genoom. Het mitochondriale genoom of mtDNA kan daarom gebruikt worden om stambomen te herleiden. Het zijn de eicellen die de mitochondriën van het toekomstige organisme bevatten. Deze zijn perfect afgestemd op het DNA van de nucleus. Nick Lane, een expert op het gebied van mitochondriën, beweert dat dit de reden is waarom er bijna uitsluitend twee geslachten bestaan in de planten- en dierenwereld. Eén geslacht, meestal de moeder, draagt behalve de (nucleaire) genen bij aan de mitochondriën en het andere geslacht voegt uitsluitend het vaderlijke deel van de genen toe. Een groot probleem bij kloneren is het feit dat de mitochondriën van de ontvangende cel helemaal niets te maken hebben met het DNA van denucleus. De celkern wordt immers geïnjecteerd in de eicel. Het is dan te verwachten dat een gekloneerd dier niet oud zal worden. De keten van het elektronentransport lekt natuurlijk en zorgt voor het ontstaan van vele vrije radicalen . Gemuteerd mtDNA /bron van de afbeelding : http://www.mda.org/publications/mitochondrial_myopathies.html(het is een site over mitochondrial myopathies): Nu is het mitochondriale genoom onderhevig aan een hoge mutatiesnelheid (3 tot 2,7 x 10-5 per generatie van 20 jaar) vergeleken met dat van de nucleus (2,5 x 10-8 per generatie van 20 jaar). Het mitochondriale DNA heeft nauwelijks reparatiesystemen van schade of andere mutaties en de mutatiesnelheid ligt dus hoger vergeleken met dat van de nucleus waar er een uitgebreid arsenaal aan reparatie-enzymen bestaat. Bovendien worden in de mitochondriën de 'fouten' makkelijker gefixeerd . Er zijn per cel immers meer mitochondriën en sommige ontbrekende functies in één mitochondrium worden eenvoudigweg getolereerd door de aanwezigheid van andere mitochondriën in dezelfde cel. Dit betekent dat het mitochondriale genoom behoorlijk uit de pas kan lopen als deze 'fouten' accumuleren. (1) Het elektronentransport, waaraan zowel het nucleaire als het mitochondriale genoom bijdragen, kan daardoor vrije radicalen produceren en de veroudering in gang zetten. Raakt een cel teveel beschadigd door al deze vrije radicalen dan wordt het proces van apoptose ingezet, ofwel een zelfmoordprogramma waarbij de cel zichzelf elimineert. Het verschil in mitochondriën maakt het onmogelijk voor verschillende soorten om tussen elkaar te paren. Het ontstaan van hybriden is daardoor uitgesloten. Het was al lang duidelijk dat mitochondriën verantwoordelijk zijn voor de energiehuishouding van de cel. Sinds een aantal jaren is het duidelijk geworden, dankzij Nick Lane, dat deze organellen ook en vooral van essentieel belang zijn bij soortvorming en veroudering. Comments (Gert korthof ).... het blijft toch een onwennige gedacht: 1000x hogere mutatiefrequentie in mitochondrieën dan in het nucleair genoom en toch bestaan er nog steeds mitochondrieën met een intact genoom? In de figuur bevatten alle mature egg cells tenminste 20% gemuteerde mitochondrieën. Dat is dus een ongezonde start voor de volgende generatie. -Waarom hebben niet veel meer mensen een mitochondriale ziekte? Of: -draagt dit bij tot de frequentie van spontane abortussen? ( Marleen )ik hoopte al dat iemand deze vragen zou stellen, want ik had er zelf ook mijn twijfels over.Mijn idee is dat de eicellen zich relatief minder vaak delen dan de somatische cellen. Deze geslachtscellen worden reeds vanaf de eerste embryonale stadia aangelegd en delen zich daarna niet meer. De mitochondrien van deze cellen hoeven zich dus ook niet voortdurend te delen en zijn relatief minder onderhevig aan mutaties. Zou dit zo kunnen zijn? Ik hoop het maar want anders hebben we inderdaad veel mutaties in het DNA van de mitochondrien in de eicellen en is het moeilijk te veronderstellen dat mito’s en celkernen van eicellen goed op elkaar afgestemd zijn, zoals in de geciteerde artikelen beweerd wordt. (gebaseerd op een blog van Ed Yong, die twee artikelen van Nick Lane behandelt.) Gert Korthof in: THE ORIGINS, PATTERNS AND IMPLICATIONS OF HUMAN SPONTANEOUS MUTATION / James F. Crow wordt uitgelegd dat voor het aanmaken van sperm veel meer delingen nodig zijn dan voor ei- cellen Volwassen sperm heeft 36 delingen achter de rug tegen 24 delingen voor the egg cell. Ik ben een serie aan het bloggen over mutatie accumulatie in de mens. Ik had mutaties in mitochondrieen nog niet gedaan, dus dit hier vormt een goede aanvulling. Mutaties in mitochondrieen zijn betrokken bij bipolar disorder, soms autism, en nog een hele serie andere ziektes: http://en.wikipedia.org/wiki/Mitochondrial_disease Uit: NotRocketScience (Ed Young), Critics on Nick Lane's: Power, Sex and Suicide, DecodedScience (met dank aan ingStHawk), W.M. Brown et al. bacterien, DNA, elektronentransport, elektronentransportketen, endosymbionten, endosymbiose, genoom, LUCA, mitochondrien, veroudering, vrije radicalen evolutie, VK-bloggers, Wetenschap *Recent onderzoek wijst uit dat de laatste universele gemeenschappelijke voorouder (LUCA) wel eens complexer zou hebben kunnen zijn dan tot nu toe werd aangenomen en dat deze reeds mitochondriën bezat die in een tweede moment verloren werden door de toekomstige bacteriën. Mutatiesnelheden MtDNA http://www.plosbiology.org/article/info%3Adoi%2F10.1371%2Fjournal.pbio.0060204 Mitochondriën zijn de energie-producerende organellen van de cel, en ze bevatten genetische informatie gecodeerd op hun eigen genoom. Mitochondriaal DNA (mtDNA) varianten worden veel gebruikt in de evolutionaire genetica en wel als verklikkertjes lichtjes bij het inschatten van de geschiedenis van de divergerende soorten in verschillende taxa. M.a.w. Mitochondriale genetische variatie tussen populaties en soorten wordt veel gebruikt in dating evolutionaire gebeurtenissen en de(geografische ) verplaatsingen binnen een populatie Omdat de mutatiesnelheden van mitochondriële genomen ,(verondersteld ) hoger zijn dan die van nucleaire genomen zijn genetische verschillen tussen en binnen soorten (en hun mitochondrien )vooral nuttig in populatiegenetica, bijvoorbeeld als verklikkers van populatiebewegingen Een species -geschiedenis wordt gewoonlijk afgeleid van fylogenetische vergelijkingen en analyses , die tevens worden gedacht als zijnde gebaseerd op moleculaire klokken .... Monsters afkomstig van verschillende tijdstippen uit een 200 generatie durende stamlijn van Drosophila Melanogaster , zijn eveneens vergelijkend gebruikt om de mutatie snelheid in te schatten met behulp van loci die verondersteld neutraal evolueren. We waren in staat direct de mutatie snelheid te schatten in het mitochondriaal genoom van de fruitvlieg Drosophila melanogaster en wel in de stamlijnen waar willekeurig mutaties zich ophoopten en bij het vrijwel ontbreken van een effectieve natuurlijke selectie. We zochten naar nieuwe mutaties door het vergelijken van het DNA van de verschillende lijnen door een gevoelige mutatie detectie techniek toe te passen . Een schatting van de mtDNA mutatie snelheid werd bekomen door het direct scannen van mitochondriale genomen monsters van Drosophila melanogaster uit de stamlijnen die ongeveer 200 generaties duurden en een spontane mutatie accumulatie (MA) vertoonden . In totaal werden 28 puntmutaties en acht insertie-deletie (indel) mutaties, gedetecteerd , waardoor een raming voor de mutatie -snelheid van een enkelvoudige -nucleotide mutatie(= een punt-mutatie ) aan een snelheid van 6,2 × 10 -8 per locus / per vliegen generatie , werd bekomen . De meeste mutaties werden heteroplastisch binnen de stamllijn, en hun frequentie verdeling suggereert dat het effectieve aantal van de mitochondriale genomen ,overgedragen per vrouwelijke stamlijn /per generatie, ongeveer op 30 ligt We observeerden herhaaldelijk ook het voorkomen van indel mutaties, wat suggereert dat indel mutatie-hotspots aanwezig zijn. Onder de puntmutaties, is er een grote hoeveelheid van het G → A type en dat op de grote streng (de sense streng voor de meerderheid van de mitochondriale genen). Deze mutaties hebben de neiging op te treden in de nonsynonieme sequenties van eiwit-coderende genen, en ze zijn schadelijk( zoals verwacht ) , ze worden dus niet geconserveerd We laten zien dat de mitochondriale mutatie snelheid , ongeveer tien keer hoger ligt dan de nucleaire DNA-mutatie snelheid . De algehele mtDNA mutatie snelheid , per basepaar / per vliegen generatie in Drosophila , wordt geschat op ongeveer 10 x hoger dan de nucleaire mutatie snelheid, maar de belangrijkste mitochondriale "G → A "mutatie snelheid is ongeveer 70 x hoger dan die van nucleaire mutatie-sdnelheid . " Silent sites" neigen aanzienlijk sterker in de richting A en T dan nonsynonieme sites, wat in overeenstemming is met de extreme mutatie voorkeur voor A + T. Streng -asymmetrische mutatie bias, in combinatie met de selectie en conservatie van specifieke nonsynonieme bases , biedt dan ook een verklaring voor de extreme "default" samenstelling van het gemiddelde mitochondriaal genoom van Drosophila. Opvallend is echter, dat bijna alle van de "punt mutaties " ( enkerlvoudige -base mutaties )die we ontdekten , bestonden uit een verandering van G naar A op een aminozuur -plaats van een eiwit-coderende gen sequentie De verklaring lijkt te zijn dat natuurlijke selectie de nucleotide G op de aminozuur locus uiteindelijk conserveert , terwijl de meeste "stille "sites van de sequenties onder zwakkere selectie staan en eerder muteren naar A of T. De mutatie snelheid voor de "G naar A veranderingen" is 70 maal hoger dan de nucleaire DNA-mutatie snelheid . De extreme "gerichtheid " handhaaft de hoge A + T inhoud van het Drosophila mitochondriaal genoom. http://en.wikipedia.org/wiki/Mutation_rate Human mitochondrial DNA has been estimated to have mutation rates of ~3× or ~2.7×10 −5 per base per 20 year generation (depending on the method of estimation)[5]; these rates are considered to be significantly higher than rates of human genomic mutation at ~2.5×10−8 per base per generation[1]. 1.^ a b c Estimate of the Mutation Rate per Nucleotide in Humans M. W. Nachman, S. L. Crowell, Genetics 156, 297-304, 2000 5.^ Estimation of Past Demographic Parameters From the Distribution of Pairwise Differences S. Schneider, L. Excoffier, 1999 Kommentaren Ing st Hawk en Marleen Veiligheidsklep ? G-C’s muteren meer dan A-T’s? Mitochondrien vangen mutaties opvangen die anders het nucleaire DNA beschadigen? De mutatiesnelheid van mtDNA ligt bij de fruitvliegjes duidelijk lager dan die bij de mens. Er bestaan grote verschillen in mutatiesnelheid tussen soorten wat volgens Lane (zieYong) te maken heeft met de grootte van het nest. Weinig jongen betekent een lage mutatiesnelheid (en de mens dan?). Ook dieren die een snel en efficient metabolisme (nodig) hebben, zoals vogels en wellicht ook fruitvliegjes hebben een lage mutatiesnelheid van het mtDNA. Muller's Ratchet Knelpunten(bottle- necks ) in (asexuele) kiembanen en de evolutionaire conservatie van mitochondriale genomen. Genetics 149: 2135-2146 http://pritch.bsd.uchicago.edu/publications/pdfs/BergstromEtAl98.pdf. *Verschillende functies van mitochondria suggereren dat mitochondria gevoelig kunnen zijn voor Muller's ratchet en andere vormen van evolutionaire degradatie:( =genetic meltdown(?) (1) in asexuele soorten ? ) Mitochondriën zijn voornamelijk uniparentaale in hun afstamming , lijken non-recombinant , en hebben een hoge mutatie frequentie met aanzienlijke aantallen schadelijke en licht schadelijke varianten . Het niettegenstaande deze "degeneratie " toch blijvend voortbestaan van mitochondria kan worden verklaard door gebruik te maken van recente gegevens die wijzen op reeds in hun evolutionaire geschiedenis genomen ernstige " knelpunten"(= bottle necks ) voor een aantal mitochondriën in de geslachtscellen bij de mens en andere zoogdieren. *We stellen een populatie-genetisch model voor , waarbij schadelijke mutaties zich voordoen binnen de afzonderlijke mitochondriën, terwijl de natuurlijke selectie werkt op assemblages(constellaties / netwerken ) van mitochondriën op het niveau van hun eukaryotische gastheren. *We tonen aan in de studie dat een "bottle neck "de werkzaamheid van de natuurlijke selectie tegen schadelijke mutaties laat toe nemen en wel door het verhogen van de variatie in fitness onder eukaryotische gastheren. *We onderzochten zowel de evenwichte verdeling van de schadelijke en licht schadelijke mutaties in grote populaties, als ook de dynamiek van Muller's Ratchet in kleine populaties. *We bevonden dat in de afwezigheid van de MR een bottle neck leidt tot betere mitochondriale prestaties over een langere tijdschaal, een "bottle neck " vertraagt de degenariever accumulatie van de MR (1) Bestaat er een ‘genomic melt-down’? (Gerdien De Jong ) ‘Genomic meltdown’ ......bestaat volgens Google alleen op de site theologyweb (en welke richting daar bovendrijft is niet moeilijk raden). Muller’s ratchet http://en.wikipedia.org/wiki/Muller's_ratchet Creationist PB (comments ) http://www.sterrenstof.info/?p=1592 Ik was deze week op het SystemsX congres http://conference.systemsx.ch/ "....DR Vamsi Mootha, Harvard, USA vertelde dat hij de complete mtDNA genomen had gesequences van 40 patienten en 40 controles. Hij wilde namelijk wel eens weten welke mutaties de door hem bestudeerde ( mitochondriale ? ) ziekten veroorzaken. Tot zijn grote verbazing ( althans dat beweert PB ) trof hij evenveel degeneratieve mutaties aan in de mitochondrieen van gezonde mensen als in die van de zieken! Dit waren de resultaten: In de zieken trof hij 17 zelzame degeneratieve allelen aan, in de gezonden 14 ( en PB trekt onmiddelijk de hem welgevallige "conclusie " ) Waarom de gezonden gezond zijn komt door de bufferende capaciteit van de netwerken waarin ze opereren. Darwins selectiemechanisme is werkelijk helemaal onderuit gehaald door de bevindingen van de moleculaire biologie. Mensen als Fransen en De Jong zijn NeoDarwinisten. Dat is een 19e eeuwse materialsitische filosofie en ze proberen die te verenigen met een theologie. Maar dat gaat niet. Dàt willen ze niet inzien. Maar er is geen enkele reden om de twee te verenigen of met elkaar in overeensteming te brengen, want de biologie is helemaal niet DArwinstisch verklaarbaar. ( Gerdien ) =maar die voorkomende mutaties en degeneratieve gevolgen zijn allemaal een bekend verschijnsel bij haploide asexuelen (zoals ook mitochondria dat zijn ). Uiteraard is een mtgenoom niet hetzelfde als een diploid nucleair genoom Muller's ratchet - blijft beperkt tot asexuelen. De bekendste verklaring voor het bestaan van sexuele voortplanting (= Muller's ratchet theory ) is dat MR het mogelijk maakt te ontsnappen aan Muller’s ratchet, dus mogelijk maakt licht schadelijke mutaties kwijt te raken. Dat wil zeggen dat ‘genomic meltdown’ niet zo gemakkelijk is bij sexueel voortplantende organismen. Attachment: Crow.JF_NatRevGen_00.pdf spontaneous mutations.pdf Attachment: BergstromEtAl98.pdf http://home.kpn.nl/b1beukema/mitochondrien.htm Ik mocht volgend zeer welkom E- mailtje ontvangen van BENNO BEUKEMA "Bij deze wil ik u wel toestemming geven om gedeelten van de inhoud te gebruiken zolang dat maar met een verwijzing naar de oorspronkelijke pagina gebeurt." Benno Beukema wat ik bij deze ook graag doe http://home.kpn.nl/b1beukema/mitochondrien.html ik vewijs verder graag naar de komplete prachtige site vol informatie van B. Beukema Mitochondriën: Enkelvoud: mitochondrion ( Grieks van "mito" en "khóndrion" ). Ook wel: mitochondrium ( verlatijnste vorm), meervoud is dan mitochondriën of mitochondria. Mitochondriën zijn celorganellen die de in koolhydraten en vetten aanwezige energie overdragen aan ATP en zo ter beschikking stellen van energievragende reacties in de cel. Het mitochondrion werd in 1886 voor het eerst gezien door Richard Altmann (Duits histoloog; 1852- Leipzig 1901), die dacht dat deze organellen intracellulaire parasieten waren. (Hij observeerde verschillende granula in het cytoplasma van diverse cellen. Hij dacht dat deze granula "elementaire organismen" konden zijn die in het cytoplasma leefden en noemde deze "bioblasts"). In 1897 vormde Carl Benda ( geboren op 30-12-1857 in Berlijn en overleden op 24-05-1932 in Turijn ) het woord mitochondrion uit " mito " ( schroefdraad) en " khóndrion " ( korreltje ), omdat deze organellen onder een lichtmicroscoop op schroefdraadvormige korreltjes lijken. Mitochondrion Korte historie: 1774 Joseph Priestly en Antoine Lavoisier, ontdekken zuurstof en de ademhalingsketen 1857 Rudolph Kölliker, pionier van de lichtmicroscoop, ziet mitochondriën in spieren. 1890 Richard Altmann, ontwikkelt mitochondriële kleuring, postuleert genetische autonomie. 1897 Carl Benda bedenkt naam uit "mito" en "khóndrion " 1912 Otto Warburg, identificeert ademhalingsketen enzym, ruwe isolatie mitochondriën. 1923-1933 David Keilin, identificeert cytochromen en de redox keten. 1940-1943 Albert Claude, isoleert intacte lever mitochondriën en microsomen 1948-1951 Albert Lehninger, lokaliseert β-oxidatie, TCA en oxphos in mitochondriën 1952 George Palade, gebruikt EM om de binnenste- en buitenste membranen en cristae te definiëren. 1950's Britton Chance, regeling van mitochondriële ademhalingsketen. 1961 Peter Mitchell, chemieosmose theorie 1972 Denham Harman, mitochondriële vrije radicalen theorie betreffende het ouder worden 1974 David Nicholls, mitochondrial proton leak regulates BAT heat production 1981 Lynn Margulis, endosymbiose theorie 1981 Fred Sanger, mtDNA sequence 1986 Kay Tanaka en collega's, eerste mtDNA ziekte gerapporteerd 1996 Xiaodong Wang, reports cytochrome c release central to apoptosis Historie: Wetenschappers geloven dat ongeveer 1,5 miljard jaar geleden eukaryote cellen hun benodigde energie verkregen door een serie relatief inefficiënte reacties, waarbij geen zuurstof nodig was. Zuurstof, dat een afvalproduct van enkele van deze processen was, begon zich langzaam in de atmosfeer op te bouwen. Wetenschappers denken dat tijdens deze periode een primitieve eukaryote cel een primitieve bacterie heeft verslonden die het vermogen had gekregen zuurstof te gebruiken om grote hoeveelheden energie te produceren. Over miljoenen jaren begon er een symbiotische relatie te ontstaan tussen de cellen en vandaag de dag hebben alle cellen van planten en dieren organellen die de afstammelingen zijn van deze oorspronkelijke energie fabriekjes. In dierlijke cellen worden deze organellen mitochondriën genoemd. Planten hebben naast mitochondriën nog een tweede soort energie producerend organel, namelijk de chloroplast. De endosymbiose theorie: De intracellulaire organisatie van de levende cel , bestaande uit gespecialiseerde organellen, maken complexe levensvormen mogelijk. Het is onbetwistbaar , gezien de getuigenis van vele fossielen , dat eencellige organismen met weinig of geen intracellulaire organisatie eens de aarde domineerden. Bij het begin van het leven ( zoals dat gedefinieerd wordt ), regeerden blauwgroene algen de planeet , maar namen na 1,6 miljard jaar in hoeveelheid af , doordat ze zuurstof produceerden . Dit zuurstof kon bij een bepaald kritisch punt niet langer meer door de oceanen worden geabsorbeerd en begon zich op te bouwen in de atmosfeer. De blauwgroene algen maakten plaats voor andere celgebaseerde organismen die konden groeien in een zuurstofrijke omgeving. Deze nieuwe organismen markeerden de oorsprong van de eukaryote ( eukaryoot : een organisme waarvan de cellen een kern bezitten die omgeven is door een kernmembraan ) cel, die ongeveer verscheen toen het zuurstof niveau tot ongeveer 3 % van zijn huidige niveau was gestegen. (Crawford and Marsh, 1995, p. 69 ). Hoewel deze algemene chronologie als feit geaccepteerd wordt, bestaat er geen zekerheid over hoe de eukaryote cel op het toneel verscheen en, meer specifiek, hoe de eukaryote mitochondrion op het toneel verscheen. Darwinisten, of voorstanders van de natuurlijke selectie theorie, zullen argumenteren dat de blauwgroene algen muteerden , waarbij over een periode van miljoenen jaren selectieve gunstige mutaties werden gemaakt, totdat door competitie en selectie een winnende combinatie de algen ( als een andere levensvorm ) in staat stelden te overleven in een zuurstofrijke omgeving. Anderen geloven dat symbiose, en meer specifiek de endosymbiose, het begin van de eukaryote cel was. Deze theorie die voorgestaan wordt door Lynn Margulis in haar boek “Symbiosis in Cell Evolution “ uit 1981 bepleit de volgende chronologie: 1. Blauwgroene algen produceerden zuurstof als een bijproduct van de fotosynthese , waarbij het zuurstof in staat werd gesteld zich op te bouwen in de atmosfeer. 2. Naast de blauwgroene algen ontwikkelden zich andere bacteriën waarvan sommige aerobische ( zuurstof gebruikende ) mogelijkheden hadden. 3. Anaërobische, heterotrofische cellen ( proto eukaryote cellen ) namen deze cellen in of verzwolgen deze aërobische bacteriën en ontwikkelden een gezamenlijke nuttige relatie. Hoe konden beide soorten daar voordeel uit halen ? De ingenomen aërobische bacterie ontving voedsel van de gast , terwijl de gast energie verkreeg door de aërobische activiteit van de bacterie. Hoewel er vele toepassingen zijn van de symbiose theorie van de cel evolutie , is het onderwerp van deze discussie het ontstaan en de functie van de mitochondriën in eukaryote cellen. In het hierboven gestelde scenario was de aërobische bacterie die door de anaërobische bacterie was ingenomen , een proto mitochondrion. Met andere woorden , een organisme die het mogelijk maakte energie te produceren uit zuurstof. Symbiose , de relatie waarbij beide soorten een gezamenlijk voordeel behalen, zoals hierboven beschreven, wordt door sommigen beschouwd als het proces waarbij mitochondriën organellen werden in eukaryote cellen. Voordat we verder gaan graven in het bewijs voor deze theorie, moeten we eerst eens kijken naar de functie en structuur van de mitochondriën zoals we die vandaag de dag in onze cellen aantreffen. Mitochondriën zijn eukaryote organellen die de oxidatieve ademhaling uitvoeren, de laatste stap in de cellulaire ademhaling. De oxidatieve ademhaling breekt pyruvaat af die in de glycolyse wordt gevormd waarbij kooldioxide wordt gevormd en produceert de grootste hoeveelheid ATP in de cellen. In eukaryote cellen is zuurstof nodig , omdat mitochondriën zuurstof gebruiken als de uiteindelijke elektronen acceptor in de elektronen transportketen, die tenslotte resulteert in een protonen gradiënt die de ATP synthese aandrijft. Mitochondriën zijn in verschillende hoeveelheden aanwezig in verschillende eukaryote cellen . Cellen die veel energie nodig hebben zoals spierweefsel en de lever hebben verhoudingsgewijs meer mitochondriën dan bijvoorbeeld cellen die minder energie nodig hebben zoals botweefsel. Hoe is het mitochondrion opgebouwd? De kenmerken die we hier bekijken zijn de omvang van het mitochondrion, de membraan structuur, proteïnen status en genetische informatie. Mitochondriën zijn een van de grootste organellen in eukaryote cellen met een omvang van 0,3-1,0 micrometer tot 5-10 micrometer. Het heeft twee membranen waarvan de binnenste sterk geplooid is en cristae worden genoemd. De cristae zijn het startpunt voor enzymen en elektronendragers ( cytochromen ) die verantwoordelijk zijn voor de elektronen transport en de oxidatieve fosforylering. Vergeleken met alle andere organellen in de cel zijn mitochondriën uniek omdat ze hun eigen DNA bevatten, dat wil zeggen , gescheiden van het DNA in de celkern. Enkele proteïnen van het mitochondrion worden door ribosomen in het mitochondrion geproduceerd , overeenkomstig zijn eigen onafhankelijk DNA. Wat is nu het bewijs die de endosymbiose theorie van het mitochondrion en de eukaryote cel ondersteunt? Enkele van de meest overtuigende bewijzen van de symbiose theorie werd hierboven reeds genoemd. Indien mitochondriën eens vrij levende bacteriën waren, bestaat de mogelijkheid dat er overblijfselen van hun vroegere conditie zijn overgebleven , ondanks dat ze vandaag de dag organellen zijn. Hieronder zullen we 6 punten bespreken: 1. In het algemeen genomen hebben mitochondriën en bacteriën dezelfde grootte. Dit kan niet worden gezegd van de andere organellen in de eukaryote cel. 2. Zoals bacteriën hebben ook mitochondriën dubbele membranen. De samenstelling van de lipiden van de membranen laten geen overeenkomsten zien met het cytoplasma van de eukaryote cel. Indien mitochondriën evolueerden binnen een proto eukaryote cel , dan zou men verwachten dat de samenstelling van de membranen uit het zelfde materiaal opgebouwd zou zijn. Het blijkt echter dat de samenstelling van de mitochondriële membranen , wat de samenstelling van lipiden betreft , meer op bacteriële membranen lijken. (Margulis, 1981, p. 217). 3. Ook de sterke plooien van de binnenste membraan bepleit voor de symbiose theorie. Volgens Margulis zijn de plooien aanpassingen die als doel het vergroten van de oppervlakte hebben voor de oxidatieve enzymen.; analoog aan de evolutie van mesosomale membranen van vele prokaryote cellen. (Margulis, 1981, p. 208). Verder houden de plooien de verschillende enzymen gescheiden overeenkomstig het gebruik, net als bacteriën doen. 4. De mitochondriële ribosomale RNA reeks heeft meer weg van bacteriën dan van ribosomen in het eukaryote cytoplasma. Bijvoorbeeld : , n-formylmethionyl transfer RNA wordt alleen gevonden in mitochondriën en bacteriën. (Dyer and Obar, 1985, p. 78). 5. Niet alleen bevatten mitochondriën hun eigen DNA, maar het is ook cirkelvormig, net als het DNA van bacteriën. Verder is de verhouding van guanine-cytosine base pairs in het mitochondriële DNA verhoudingsgewijs hoger , net als bacteriën, dan als het DNA in de celkern. (Margulis, 1981, p. 206). 6. Volgens Margulis lijkt de deling van mitochondriën meer op de reproductie van bacteriën. Zij schrijft : "Genetic recombination in (mitochondria) is far more reminiscent of phage and bacterial sexuality than it is of eukaryotic nuclear sexual behavior" (Margulis, 1981, p. 218). Meer ondersteunend bewijs voor de symbiose theorie volgt uit het feit dat mitochondriële ribosomen vergelijkbare antibiotische gevoeligheden hebben met bacteriële ribosomen dan met eukaryote ribosomen. Bijvoorbeeld : cycloheximide blokkeert eukaryote ribosomen door de uitwerking op de overdracht van tRNA, maar het heeft geen uitwerking op mitochondriën en bacteriën. Aan de ander kant zullen stoffen die de prokaryote synthese blokkeren, maar niet de eukaryote synthese, ook de mitochondriële synthese blokkeren, bijvoorbeeld erythromycine en tetracycline (Margulis, 1981, p. 217-218). De structurele overeenkomsten tussen mitochondriën en bacteriën zijn overweldigend , maar zeker niet afdoende. Velen betwijfelen de mogelijkheid van de symbiose theorie …. Dat er een vrij levende proto mitochondrion bestond ten tijde van de opbouw van zuurstof in de atmosfeer …, dat het op de een of andere manier een proto eukaryote cel binnendrong…., dat het een partner was in een symbiotische relatie die uiteindelijk resulteerde in een proto mitochondrion, daarbij zijn autonomie aan een grotere cel gevend en de weg vrij makend voor de eukaryote cel….. wordt dit te veel? Omdat evolutie in vele opzichten de historie is van verschillende chemische reacties van de formatie van de aarde tot de biochemische reacties in levende cellen , zullen we de mogelijkheid van de symbiose theorie eens bekijken. Ten eerste is het niet ondenkbaar dat vrij levende aërobische bacteriën die hoge energie moleculen zoals ATP produceren , een relatie aangaan waarbij die energie kan worden gebruikt. Ten tweede moet de productie van al die energie een enorme input van energie hebben vereist. Bijvoorbeeld een voldoende, beschikbare en efficiënte voedselbron. Ten derde, de komst van zuurstof in de atmosfeer, dat een giftig gas was voor de meerderheid van de organismen op dat moment op aarde, vereiste een nieuwe vorm van stofwisseling , gebaseerd op een nieuwe chemie. Ten slotte zouden proto eukaryote cellen die geen mogelijkheid hadden zuurstof in de atmosfeer om te zetten , moeilijk kunnen overleven. De basis voor de symbiose is duidelijk : anaërobische organismen leverden een constante bron van voedsel en fosfolipiden voor zowel de mitochondriële membranen als voor bacteriën. (Crawford and Marsh, 1995, p. 71-72). Als tegenprestatie stelde de energie die vrijkwam bij de ademhaling van zuurstof, de gast in staat te overleven en zich verder aan te passen aan de nieuwe condities op de aarde. De symbiose theorie wordt gestaafd door natuurlijke , geobserveerde voorbeelden van symbiotische relaties. Bepaalde zeevissen zijn in staat licht te emitteren vanwege de aanwezigheid van lichtgevende bacteriën in hun binnenste. Deze lichtgevende bacteriën leven ook vrijelijk in zeewater, maar geven dan geen licht. (Dyer and Obar, 1985, p. 127). Andere voorbeelden van symbiose zijn de relaties tussen verschillende schimmels en cyanobacteriën, algen en planten en bacteriën en zoogdieren. (Margulis, 1981, p. 165). Hoewel de transformatie van deze symbionten een enorme stap is , als deze al heeft plaatsgevonden, vond deze wel plaats over een periode van miljoenen jaren. Om de hierboven genoemde redenen kan men de theorie, dat mitochondriën evolueerden uit vrij levende aërobische bacteriën tot organellen in de eukaryote cel, niet negeren. De vraag blijft echter , hoe en uit wat konden proto mitochondriën evolueren naast blauw groene algen? Op welke manier drongen de proto mitochondriën de grotere aërobische cel binnen? Konden voorbeelden van vrij levende proto mitochondriën overleven? Bestaan zij of hun nakomelingen vandaag de dag nog? Zijn andere eukaryote organellen ook afgeleid van vrij levende organismen ? Vanwege de bewijzen kan men de symbiose theorie niet negeren. REFERENCE CITATIONS 1. Crawford, Michael and David Marsh, 1995. Nutrition and Evolution, p. 65-83. Keats Publishing, Inc., New Canaan, CT. 2. Dyer, Betsey Dexter and Robert Obar (editors), 1985. The Origin of Eukaryotic Cells, Van Nostrand Reinhold Company, Inc., NY. Papers by: a.) Goksøyr, J., 1967, "Evolution of Eucaryotic Cells;" b.) Schwartz, R.M. and M.O. Dayhoff, 1978, "Origins of Prokaryotes, Eukaryotes, Mitochondria, and Chloroplasts;" c.) Raven, P.H., 1970, "A Multiple Origin for Plastids and Mitochondria;" d.) Doolittle, W.F., 1980, "Revolutionary Concepts in Evolutionary Cell Biology;" and e.) Smith, D.C., 1979, "From Extracellular to Intracellular: The Establishment of a Symbiosis." 3. Margulis, Lynn, 1981. Symbiosis in Cell Evolution, p. 206-227. W. H. Freeman and Company, San Francisco. 4. Prescott, L.M., J.P. Harley and D.A. Klein, 1996. Microbiology, third edition. Wm. C. Brown Publishers, Dubuque, IA Het mitochondrion: De uitwendige vorm van mitochondriën is variabel; van ongeveer bolrond tot zeer langwerpig. Mitochondriën zijn na de celkern de grootste organellen in dierlijke cellen. De mitochondriën variëren in diameter van 0,5 tot 1 micrometer en kunnen tot 7 micrometer lang zijn. ( 1 micrometer is 1 miljoenste meter ). Mitochondriën zijn nog juist onder de lichtmicroscoop herkenbare lichaampjes, omgeven door een dubbele membraan . Beide membranen zijn 4–6 nanometer dik, met een tussenruimte van 6–9 nm. ( 1 nanometer is 1 miljardste meter ) . Beide mitochondriële membranen hebben belangrijke verschillen in hun samenstelling. De buitenste membraan bevat een transport proteïne die porin wordt genoemd. Daardoor wordt de buitenste membraan doorlaatbaar voor alle moleculen die kleiner of gelijk zijn dan 5000 Dalton ( een eenheid van moleculair gewicht ; Hierbij is 1 dalton gelijk aan 1 atomaire massa-eenheid en deze is weer gedefinieerd als 1/12 van de massa van een neutraal 12C atoom.). Porine ( VDAC ) is een proteïne die watergevulde transmembrane kanalen vormt, waardoor ionen van niet specifieke moleculen het membraan kunnen passeren. Ook bevat dit membraan proteïnen die substraten van lipiden om kunnen zetten in vormen die gebruikt kunnen worden in de matrix. De ruimte tussen de binnenste en buitenste membranen ( intermembrane space ) bevatten enzymen die ATP gebruiken om andere nucleotiden te fosforyleren. De binnenste membraan bevat een hoog gehalte aan cardiolipin , waardoor de binnenste membraan ondoorlaatbaar is voor ionen. Cardiolipine is een fosfolipide : diphosphatidyl glycerol . De binnenste membraan is permeabel voor O2 , CO2 en H2O. Verder bevat dit membraan specifieke transport proteïnen voor moleculen die nodig zijn voor de mitochondriële enzymen in de matrix ( ATP, ADP , pyruvate , Ca2+ en fosfaat ). De binnenste membraan bevat de proteïnen die de oxidatieve reacties uitvoeren van de ademhalingsketen. Ook bevat dit membraan het complex ATP synthetase die de ATP maakt. De binnenmembraan is sterk geplooid, de plooien (cristae mitochondriales) staan vaak ongeveer loodrecht op de wand. Het doel van deze plooien is een veel groter oppervlak te creëren , waardoor meer werk kan worden verricht in een kleine ruimte. De matrix ( de ruimte binnen de binnenste membraan ) is de plaats waar de citroenzuurcyclus plaatsvindt , verder bevat deze ruimte DNA , speciale mitochondriële ribosomen , tRNA en verschillende enzymen die nodig zijn voor het mitochondriële gen. Elektron Microscopic Atlas : Mitochondriën Ze hebben een autonoom karakter: ze bevatten DNA, beschikken over een eigen RNA-synthese en bezitten ribosomen (die sterk overeenkomen met ribosomen van bacteriën). Mitochondriën zwellen op en krimpen in als respons op verschillende hormonen en drugs en tijdens de synthese van ATP. Dit opzwellen en inkrimpen lijkt gerelateerd te zijn aan de beweging van water door de cellen. Mitochondriën kunnen zich, evenals de kern en de plastiden, alleen vermeerderen door deling. Mitochondriën delen zich op een manier die morfologisch lijkt op de manier zoals bacteriën zich delen. De aantallen mitochondriën worden geregeld door autofagie; dit is een proces waarbij de lysosomen betrokken zijn. Mitochondriën bevatten o.a. de enzymen van de citroenzuurcyclus en van de ademhalingsketen (eindoxidatie), inclusief die voor de vorming van het energierijke ATP en ook die voor de afbraak van vetzuren. Een deel van deze enzymen is aan de cristae mitochondriales gebonden. Mitochondriën kunnen vele verschillende vormen aannemen en dienen voor een groot aantal verschillende functies. Elke vorm van het mitochondrion is karakteristiek voor de cel waarin het verblijft. Er zijn ongeveer 250 verschillende typen cellen in ons lichaam. Er zijn verschillende mitochondriën met specifieke metabole functies voor veel van de 250 typen cellen in ons lichaam. Mitochondriën zijn in staat hun structuur aan te passen om tegemoet te komen aan veranderende omstandigheden van de cel. Ook kan er in bepaalde gevallen een toename van de cristae plaatsvinden, of een verandering van de vorm van het mitochondrion die resulteert in een groter actief oppervlak voor de omzetting van energie. Een mitochondrion kan versmelten met een andere mitochondrion, of kunnen in grootte toenemen , waarbij " giant mitochondria " of " megamitochondria " worden gevormd. Er wordt een toename van het aantal mitochondriën gegenereerd in situaties van hoge metabole activiteit. Aantallen mitochondriën kunnen afnemen door pycnose ( degeneratieve verdichting van de celkern ) , door opzwellen of door de formatie van autofagolysosomen. Men denkt dat de levensduur van mitochondriën tussen 5 en 12 dagen ligt. Voor de biogenese van mitochondriën is het noodzakelijk dat enige honderden genen gecoördineerd tot expressie komen. De meeste mitochondriale componenten worden gecodeerd in de kern, slechts enkele op het mitochondriale DNA. De meeste cellen met een celkern bevatten in het cytoplasma ( protoplasma of vloeibare gedeelte van de cel) 500 tot 2000 mitochondriën . Sommige cellen hebben maar een paar mitochondriën. Bloedplaatjes hebben maar twee tot zes mitochondriën. Rode bloedlichaampjes hebben geen mitochondriën .Grotere aantallen zijn er voornamelijk te vinden in de organen in het lichaam die veel energie nodig hebben , zoals het hart, het brein,de lever ,de skeletspieren, inwendige klieren en het beenmerg. Dit zijn dan ook de orgaansystemen die in het algemeen betrokken zijn bij de mitochondriële ziekten. De gedachte dat mitochondriën alleen als energiefabriekjes voor de cel fungeren is een misvatting. Er zijn ongeveer 3000 genen nodig om een mitochondrion te maken. Het mitochondriële DNA codeert maar voor 37 van deze genen. De resterende genen worden gecodeerd in de celkern en de resulterende proteïnen worden naar het mitochondrion getransporteerd. Enkele algemene kenmerken van het mitochondriële DNA (mtDNA): 2 tot 10 kopieën in elke mitochondrion mtDNA codeert voor 37 genen. - codeert voor 13 subeenheden die zich alle 13 in de mitochondriële ademhalingsketen complex bevinden. - 22 tRNA genen die verantwoordelijk zijn voor de genetische code - 2 rRNA genen ten behoeve van de mitochondriële ribosomen Mitochondriële DNA variatie: - heteroplasmie: een individuele cel bevat verschillende mtDNA populaties. ( kan voorkomen bij enkele mtDNA mutaties of vanwege de aanwezigheid van meerdere mitochondriën in een enkele cel die elk verschillende mtDNA kopieën bevat). - homoplasmie: gelijksoortige mtDNA's Mitochondrial DNA (mtDNA): General features ( Neuromuscular Disease Center, Washington University, St. Louis, MO USA ) Maar ongeveer 3 % van de genen die nodig zijn om een mitochondrion te maken ( 100 van de 3000 ) zijn toegewezen om ATP te maken. Meer dan 95 % ( 2900 van de 3000 ) zijn betrokken bij andere functies gebonden aan de speciale taken van de cel waarin het mitochondrion verblijft. Deze taken veranderen naarmate ons lichaam zich ontwikkeld van embryo naar volwassene en onze weefsels groeien en zich aanpassen aan onze omgeving. Deze andere , niet ATP gerelateerde functies, zijn innig betrokken bij de meeste belangrijke metabole paden die door de cel gebruikt worden, om zijn moleculaire bouwsteentjes op te bouwen , af te breken of te recyclen. Cellen kunnen zonder mitochondriën geen RNA en DNA maken die ze nodig hebben om te groeien en te functioneren. De bouwstenen voor RNA en DNA zijn purinen en pyrimidines. Mitochondriën bevatten de "rate-limiting" enzymen van de pyrimidine biosynthese en haem synthese die nodig zijn om hemoglobine te maken. Mitochondriën in de lever zijn gespecialiseerd om ammoniak in de ureum-cyclus om te zetten. Mitochondriën zijn ook nodig voor de cholesterol stofwisseling, voor de synthese van testosteron en oestron en voor de neurotransmitter stofwisseling. Adenosinetrifosfaat ( ATP ): ATP werd in 1929 ontdekt door de Duitse chemicus Karl Lohman. Synoniemen voor ATP: ATP; adenosine 5'-triphosphate; dATP; 5'-adenylate triphosphate; 5'-rATP; rATP; 5'-dATP; 5'-deoxyadenylate triphosphate; deoxyadenosine 5'-triphosphate; deoxyadenosine triphosphate; Adenosine 5'-triphosphate disodium salt (ATP) Bruto formule : C10H16N5O13P3 Adenosinetrifosfaat Adenosinetrifosfaat ( ATP ) is een nucleotide ( stof opgebouwd uit een stikstofrijk molecuul (( purine - of pyrimidine-base )) , een suikermolecuul en fosforzuur ) dat als co-factor bij vele enzymatische reacties in het lichaam wordt gebruikt. ATP wordt uit adenosinedifosfaat ( ADP ) gevormd bij de oxidatie van suikers, eiwitten en vetten en wordt verbruikt bij energiebehoevende processen in de cel. ATP wordt beschouwd als een zgn. energierijke verbinding, omdat bij de hydrolyse van de trifosfaatgroep energie vrijkomt die voor andere doeleinden in het metabolisme gebruikt kunnen worden. Daardoor is deze verbinding geschikt om energie te leveren bij de spiercontractie, actief transport , signaalversterking, om proteïnen te synthetiseren, of voor het kopiëren van DNA, en de opbouw van nieuwe organellen en het pompen van water door membranen en de verplaatsing van cellen. De hydrolyse van de fosfaatanhydridefunctie in ATP is een thermodynamisch gunstige reactie; de energie die hierbij vrijkomt maakt het mogelijk dat in het metabolisme een thermodynamisch ongunstige reactie toch kan plaatsvinden door deze te koppelen aan de hydrolyse van een voldoend aantal moleculen ATP. In de biosynthese worden daartoe niet-reactieve verbindingen met ATP eerst omgezet in reactieve tussenproducten die daarna vlot de vereiste reactie kunnen geven. Veel voorkomend is de omzetting door ATP van een hydroxylgroep van een alcohol of een carbonzuur in een fosfaatgroep, waarbij dan respectievelijk een fosfaatester of een reactief acylfosfaat ontstaat. De hydroxylgroep met zijn slechte vertrekkende eigenschappen wordt in beide gevallen omgezet in een goede vertrekkende groep, namelijk in het fosfaatanion. Naast de energievoorziening helpen mitochondriën ook bij de regulatie van concentraties calcium en andere elektrisch geladen deeltjes in het cytoplasma. Ook recyclen zij- en breken zij de energie in vetzuren en aminozuren af . Hoewel de mitochondriën al vanaf het jaar 1880 onderzocht worden, duurde het vele jaren voordat wetenschappers het functioneren van dit organel begrepen. Het proces waarbij de mitochondriën zuurstof gebruiken om de chemische energie , die is opgeslagen in voedsel, vrij te maken , wordt cellulaire ademhaling genoemd. ( cellular respiration ). In de vroege jaren 1900 werd ontdekt dat de biochemische reacties van dit type ademhaling in twee hoofdgroepen vallen : het koolstof traject , waarin suiker wordt afgebroken tot kooldioxide en waterstof ; en het waterstof traject waarbij waterstof in stappen wordt omgezet in zuurstof , waarbij water wordt gevormd en energie vrijkomt. In het waterstof traject passeren de elektronen van waterstof de " elektronen transportketen " . Deze elektronen transportketen bestaat uit 5 enzymcomplexen. Tijdens het transport van deze elektronen door de elektronen transportketen, geven de elektronen een deel van hun energie af. Deze energie wordt dan opgeslagen in moleculen ATP. Aan het einde van de keten zijn voor elke molecuul suiker, 38 moleculen ATP gevormd. De overall energiebalans per glucose molecuul is: Aerobisch Anaerobisch 2 ATP gebruikt in de glycolyse - 2 ATP - 2 ATP 4 ATP gevormd in de glycolyse + 4 ATP + 4 ATP 2 NADH2 gevormd in de glycolyse via elektronentransportketen + 6 ATP 8 NADH2 gevormd in de citroenzuurcyclus via de e.t. + 24 ATP 2 GTP in de citroenzuurcyclus + 2 ATP 2 FADH2 in de citroenzuurcyclus via de elektronentransportketen + 4 ATP Totaal : 38 ATP 2 ATP Soms wordt een totaal van 36 ATP aangehouden, omdat bekend is dat in eukaryote cellen het gereduceerde NAD, dat gevormd is door de glycolyse in het cytoplasma, actief getransporteerd moet worden over de mitochondriële membraan om beschikbaar te komen voor de elektronentransportketen. Het actieve transport over het membraan kost 1 ATP voor elk NADH dat getransporteerd wordt. Wanneer men zo wil redeneren , moet men ook het actieve transport van andere moleculen in ogenschouw nemen. ( pyruvaat ? , fosfaat ? , Mg+ etc ). Het juiste netto resultaat van ATP is onbekend , maar moet worden beschouwd als minder dan 36. Mitochondriën zijn verbazingwekkend efficiënt bij de omzetting van de chemische energie in suiker tot ATP. Een verbrandingsmotor wordt al efficiënt beschouwd , als het 25 procent van de chemische energie die is opgeslagen in gasolie, omzet in bewegingsenergie. Mitochondriën echter, zetten 54 procent van de energie die is opgeslagen in suiker, om in ATP. Hoewel het al lang werd vermoed werd pas in 1988 bewezen dat defecten in mitochondriële genen konden leiden tot erfelijke ziekten. In 1988 toonde Douglas Wallace van de Emory University aan dat de zeldzame ziekte " Leber's hereditary optic neuropathy " werd veroorzaakt door mutaties in het mitochondriële DNA. Het verschil tussen de mitochondriële en peroxisomale β-oxidatie: Mitochondriën oxideren korte- , midden - en grotendeels lange keten vetzuren, terwijl peroxisomen de meeste zeer lang keten vetzuren en sommige lange keten vetzuren oxideren. Mitochondriën gebruiken een acyl-CoA dehydrogenase om acyl-CoA om te zetten tot enoyl-CoA. Dit enzym transporteert de elektronen naar FAD en vervolgens naar de elektronentransportketen. Peroxisomen gebruiken een acyl-CoA oxidase welke uiteindelijk de elektronen naar zuurstof transporteert, waarbij waterstofperoxide wordt gevormd die weer door catalase wordt omgezet in water en zuurstof. Verder worden de mitochondriële en peroxisomale enzymen gecodeerd door verschillende genen. De mitochondriële vetzuuroxidatie: Het carnitine palmitoyltransferase systeem en de beta-oxidatie: De hoofdbron voor de energie productie in de cel is de mitochondriële beta-oxidatie van lang-keten vetzuren. Lang-keten vetzuren kunnen niet direct de mitochondriële membranen passeren en moeten eerst worden omgezet. Dit in tegenstelling tot de kort - keten en middellange - keten vetzuren die door diffusie de membranen kunnen passeren. De oxidatie van lang-keten vetzuren in het mitochondrion worden gekatalyseerd door de enzymen CPT I en CPT II . Samen met acyl CoA synthetase en carnitine-acylcarnitine translocase verzorgen zij het transport van vetzuren over de membranen van het mitochondrion naar de mitochondriële matrix, waar deze vetzuren uiteindelijk worden omgezet in energie in de vorm van ATP. Om de mitochondriële membranen te kunnen passeren , worden de lang-keten vetzuren eerst geactiveerd door coënzym A en dan verbonden met L-carnitine, een reactie die gekatalyseerd wordt door het enzym carnitine palmitoyltransferase ( CPTase ). Een deel van de CPTase activiteit ( CPT I ) is geassocieerd met de buitenste mitochondriële membraan en katalyseert de vorming van palmityl-carnitine. Palmityl-carnitine kan als carnitine ester de buitenste membraan passeren. Een andere activiteit van CPTase ( CPT II ) wordt geassocieerd met de binnenste mitochondriële membraan . CPT II zet in het mitochondrion palmityl-carnitine om in palmityl-CoA. Tot slot initiëren CPT II en carnitine-acylcarnitine translocase de mitochondriële oxidatie van lang-keten vetzuren. Het Carnitine Palmitoyltransferase ( CPT ) systeem, heeft betrekking op twee verschillende proteïnen, die gelokaliseerd zijn op de buitenste - en binnenste mitochondriële membranen. CPT I wordt op de buitenste mitochondriële membraan gevonden en CPT II wordt op de binnenste mitochondriële membraan gevonden. CPT II wordt in alle cellen gevonden. Van CPT I bestaan twee weefsel-specifieke isoformen, die L-CPT I en M-CPT I worden genoemd. L-CPT I wordt voornamelijk in de lever gevonden. M-CPT I wordt voornamelijk in de spieren gevonden. Het gen dat voor L-CPT I codeert, wordt CPT1A genoemd. Het gen dat codeert voor M-CPT I , wordt CPT1B genoemd. Het gen dat voor CPT II codeert, wordt CPT2 genoemd. Het gen dat codeert voor CPT I is gelokaliseerd op chromosoom 11q13. Het gen dat codeert voor CPT II , is gelokaliseerd op chromosoom 1p32. De officiële naam voor het enzym carnitine palmitoyl transferase is carnitine O-palmitoyltransferase. EC nummer 2.3.1.21 Het enzym katalyseert de volgende reactie: Palmitoyl-CoA + L-carnitine <=> CoA + L-palmitoylcarnitine Stoornissen in dit systeem leiden tot de carnitine palmitoyltransferase stoornissen. De mitochondriële β-oxidatie spiraal: De beta-oxidatie: de afbraak (dehydrogenering ) vindt plaats tussen de alfa en bèta koolstofatomen ( C2 en C3 ) in een FAD gebonden reactie, die gekatalyseerd wordt door acyl CoA dehydrogenase. De producten van de beta-oxidatie zijn acetyl CoA, FADH2, NADH en H+. Acetyl CoA is acetyl coënzym A; FADH2 is de gereduceerde vorm van flavine adenine dinucleotide en NADH is de gereduceerde vorm van nicotinamide adenine dinucleotide. De bètaoxidatie spiraal is een cyclus waarin acyl-CoA esters steeds weer de cyclus doorlopen totdat ze volledig geoxideerd zijn. Bij elke omgang in de cyclus wordt de ketenlengte met twee koolstofatomen verkort . Als voorbeeld kijken we eens naar Palmitoyl-CoA die een ketenlengte van 16 koolstofatomen heeft. De overall reactie is : 7 FAD + 7 NAD+ + 7 CoASH + H2O + H(CH2CH2)7CH2CO-SCoA > 8 CH3CO-SCoA + 7 FADH2 + 7 NADH + 7 H+ Palmitoyl-CoA maakt 7 omgangen in de bètaoxidatie spiraal door. Bij elke omgang wordt de ketenlengte twee koolstofatomen korter, waarna er uiteindelijk 8 delen met een ketenlengte van twee koolstofatomen overblijven. In de meeste weefsels zoals het hart en de spieren, worden deze 8 delen als acetyl-CoA in de citroenzuur cyclus uiteindelijk omgezet in kooldioxide en water. In de lever en de nieren wordt acetyl-CoA uit de β-oxidatie omgezet in ketonen die daarna voor de uiteindelijke oxidatie worden getransporteerd naar bijvoorbeeld het brein en de spieren. FADH2 , NADH en H+ worden geoxideerd in de elektronen transportketen waarbij ATP wordt gevormd. Elke ronde van de ß-oxidatie-spiraal wordt gereguleerd door een reeks van enzymen, waarvan sommige specificiteit bezitten voor de ketenlengte van het acyl-CoA vetzuur. Voor een verzadigd acyl-CoA vetzuur (zoals bijvoorbeeld palmitoyl-CoA ( C16 )) worden vier enzymstappen gevolgd, te weten die van: 1. 2. 3. 4. acyl-CoA dehydrogenase enoyl-CoA hydratase ß-hydroxyacyl-CoA dehydrogenase ß-ketoacyl-CoA thiolase De ketenlengte-specifieke acyl-CoA dehydrogenase enzymen zijn flavoproteïnen; in het proces waarbij een dubbele binding gevormd wordt tussen het a- en ß- koolstofatoom van een acyl-CoA-vetzuur (en waarbij dus het corresponderende enoyl-CoAvetzuur ontstaat), worden de vrijgekomen elektronen afgegeven aan ETF (elekton transfer proteïn) of ETF-dehydrogenase; dit complex legt de link tussen ß-oxidatie en ademhalingsketen. De tweede reaktie in de ß-oxidatie wordt gekatalyseerd door enoyl-CoA hydratase (crotonase). Het enoyl-CoA-vetzuur wordt hierbij gehydrateerd tot een L-ß-hydroxyacyl-CoA-vetzuur. Vervolgens katalyseert ß-hydroxyacyl-CoA dehydrogenase (bestaande uit 1 of meer enzymen) in een NAD+-afhankelijke reaktie, oxidatie van de hydroxy-groep tot een ketogroep. Bij de laatste reaktie, die gekatalyseerd wordt door ß-ketoacyl-CoA thiolase (ook weer bestaand uit 1 of meer enzymen) wordt in aanwezigheid van gereduceerd CoA, de a-ß binding verbroken. De twee produkten die bij deze reaktie ontstaan zijn acetyl-CoA, en een acyl-CoA vetzuur dat 2 koolstofatomen korter is dan het oorspronkelijke substraat. Zolang acyl-CoA-vetzuren een acetyl-CoA fragment af kunnen splitsen, kunnen ze de ß-oxidatie blijven doorlopen. Er bestaan drie acyl-CoA dehydrogenase enzymen, die ieder een verschillende ketenlengte-specificiteit bezitten; hoewel er overlap bestaat tussen deze enzymen, kan gezegd worden dat in de lever (V)LCAD ((Very) Long Chain Acyl-CoA Dehydrogenase) de eerste reaktie in de ß-oxidatie katalyseert voor acyl-CoA-vetzuren met een ketenlengte van 18 tot 12 koolstofatomen. MCAD (Medium Chain Acyl-CoA Dehydrogenase ) herkent acyl-CoA-vetzuren met een ketenlengte variërend van 14 tot 4 koolstofatomen, en SCAD ( Short Chain Acyl-CoA Dehydrogenase ) herkent alleen vetzuurketens van 4en 6 koolstofatomen. Vetzuren met een oneven ketenlengte worden ook in de bèta-oxidatie spiraal afgebroken tot er een molecuul met 3 koolstofatomen overblijft. Dit molecuul ( propionyl CoA ) kan niet verder in de bèta-oxidatie spiraal worden afgebroken, maar wordt omgezet tot succinyl CoA ( barnsteenzuur Coënzym A ) en wordt vervolgens verder omgezet in de citroenzuurcyclus. Onverzadigde vetzuren: Onverzadigde vetzuren zijn vetzuren die een of meer dubbele bindingen in hun alkyl-keten hebben. Meervoudig onverzadigde vetzuren met twee dubbele bindingen hebben normaal gesproken een dubbele binding op C9 - C10 in de alkyl-keten en een dubbele binding 3 C verder in de keten op C12- C13. De bètaoxidatie van deze vetzuren wordt doorlopen tot het punt waarop de normale enzymen van de bètaoxidatie een dubbele binding tegen komen. De enzymen van de bètaoxidatie zijn niet in staat om op substraten te reageren die een dubbele binding hebben tussen de bèta en gamma C-atomen. Om dit probleem op te lossen, verplaatst het enzym enoyl-CoA isomerase de dubbele binding een C atoom verder naar het einde van de alkyl-keten, waarna de bètaoxidatie weer een ronde verder ( en een deel van de tweede ) kan verlopen tot de volgende dubbele binding wordt aangetroffen waarna het enzym 2,4 dienoyl-CoA reductase het vetzuur reduceert waarbij een trans-3dubbele binding ontstaat. Het enzym 3,2 enoyl-CoA isomerase verplaatst deze dubbele binding verder naar het einde van de vetzuurketen, waar het vetzuur als substraat voor de normale bètaoxidatie enzymen kan dienen. Carnitine ( L-Carnitine = levo carnitine ) Historie: 1905: W. Gulewitsch en R. Krimberg ontdekken de stof carnitine in een vleesextract . 1927: Tomita en Sendju ontdekken de chemische structuur C7H15NO3 In 1948 werd een nieuwe vitamine ontdekt die essentieel was voor de groei van de gele meelworm. Men noemde deze vitamine naar de Latijnse naam van de meelworm ( Tenebro mollitor): vitamine BT. In 1952 werd vitamine BT geïdentificeerd als carnitine. 1955: ontdekking van het enzym carnitine acyltransferase. 1958: I.B. Fritz ontdekt dat carnitine de lange keten vetzuuroxidatie stimuleert in de mitochondriën. 1963: Ontdekking van het enzym carnitine palmitoyltransferase. 1973: eerste artikelen over aangeboren stoornissen in de carnitine stofwisseling. 1975: Ontdekking van het enzym carnitine acylcarnitine translocase. Ontdekking van de remming van CPT-I door malonyl CoA 1977: Ontdekking van CPT II deficiëntie met normale CPT I. 1982: Ontdekking van een carnitine transportdefect als primaire oorzaak van carnitine deficiëntie. 1987: lokalisatie van CPT I in de buitenste mitochondriële membraan. 1991: ontdekking van een fatale vorm van CPT II deficiëntie. 1992: ontdekking van carnitine acylcarnitine translocase deficiëntie. Carnitine ( 3-hydroxy-4-trimethyl aminoboterzuur : C7H15NO3 ) is een kleine, water oplosbare, quaternaire stikstof bevattende stof . Het wordt in het lichaam in de lever, het brein en de nieren gesynthetiseerd uit lysine en methionine ( met cofactoren zoals ascorbinezuur, niacine , vitamine B6 en gereduceerd ijzer ). Carnitine komt zowel in de L-vorm als in de D-vorm voor. De L- vorm ( L-carnitine ) is de biologische actieve vorm. Hoewel de mens in staat is carnitine uit lysine en methionine te synthetiseren, moet tweederde van de benodigde carnitine uit het voedsel gehaald worden. Carnitine komt met name in rundvlees, schapenvlees en lamsvlees voor. Melk en kaas bevatten wat minder carnitine, terwijl fruit en groenten maar minieme hoeveelheden carnitine bevatten. Carnitine is geen aminozuur , maar een vitamine-achtige stof. Carnitine wordt niet afgebroken , maar wordt in de urine uitgescheiden als vrije carnitine en veresterde vormen van carnitine. Carnitine functioneert in een driedelig enzymcomplex ( carnitine acyltransferase I, carnitine translocase, en carnitine acyltransferase II ) , die verantwoordelijk is voor het transport van lange keten vetzuren over de binnenste mitochondriële membraan. Naast bovengenoemde sleutelfunctie speelt L-carnitine nog een rol bij enkele andere metabole functies. Carnitine verbetert de metabole flux in de citroenzuurcyclus en voorkomt de stapeling van lange keten Acyl CoA. Verder fungeert L-carnitine als drager voor acyl-deeltjes ( verkort in de peroxisomale beta-oxidatie ) van het peroxisoom naar het mitochondrion, waar deze deeltjes verder geoxideerd kunnen worden. Carnitine is dus direct of indirect betrokken bij verschillende metabole paden en de beschikbaarheid van carnitine is een belangrijke factor die niet alleen de oxidatie van vetzuren en ketonlichamen regelt, maar ook die van glucose en andere aminozuren.. De Synthese van Carnitine in het lichaam: 1. EC 1.14.11.8 trimethyllysine dioxygenase holoenzyme homodimer trimethyllysine + alpha-ketoglutarate + O2 => beta-hydroxy-trimethyllysine + succinate + CO2 C9H20N2O2 + C5H6O5 + O2 => C9H21N2O3 + C4H6O4 + CO2 Cofactor(s): Ascorbate; Iron(2+). 2. EC 2.1.2.1 serine hydroxymethyltransferase holoenzyme tetramer beta-hydroxy-trimethyllysine => gamma-butyrobetaine aldehyde + glycine Cofactor(s): Pyridoxal-phosphate 3. EC 1.2.1.47 4-trimethylaminobutyraldehyde dehydrogenase homotetramer gamma-butyrobetaine semialdehyde + NAD+ => gamma-butyrobetaine + NADH + H+ 4. EC 1.14.11.1 gamma-butyrobetaine hydroxylase holoenzyme dimer gamma-butyrobetaine + alpha-ketoglutarate + O2 => carnitine + succinate + CO2 C7H16NO2 + C5H6O5 + O2 => C7H16NO3 + C4H6O4 + CO2 Cofactor(s): Ascorbate; Iron(2+). De verdeling van enkele enzymen in de mitochondriële compartimenten : Buitenste membraan Binnenste membraan Intermembrane space Matrix Cytochrome-b5 reductase NADH dehydrogenase (ubiquinone) Adenylate kinase [Pyruvate dehydrogenase(lipoamide)] kinase Cytochrome b5 monoamine oxidase Succinate dehydrogenase Nucleosidediphosphate kinase [Pyruvate dehydrogenase (lipoamide)]-phosphatase Kynurine hydroxylase QH2:cytochrome c oxidoreductase Nucleoside monophosphokinase Dihydrolipoamide dehydrogenase Glycerol-3-phosphate Oacyltransferase Cytochrome c oxidase Holocytochrome-c synthase Aconitase Lysophosphatidyl acyl transferase Phosphatidylserine decarboxylase Sulfite oxidase Isocytrate dehydrogenase ( NAD ) Phosphatidate phosphatase beta-hydroxybutyrate dehydrogenase Sulfite reductase Isocytrate dehydrogenase ( NADP ) Phospholipase A2 Pyridine nucleotide transhydrogenase Coproporphyrinogen oxidase Fumarase Nucleoside diphosphokinase Carnitine Opalmitoyltransferase Cytochrome-c peroxidase Glutamate dehydrogenase Porin; Voltage-dependent anion-selective channel (VDAC) Ferrochelatase Cytochrome-b5 reductase Pyruvate carboxylase Carnitine Oacetyltransferase 5-aminolevulinic acid synthase Aspartate aminotransferase 1acylglycerophosphocholine O-acyltransferase Isocitrate dehydrogenase (NADP+). Ornithine carbamyl transferase Hexokinase Pyrroline-5carboxylate reductase Carbamoyl-phosphate synthase (glutaminehydrolysing). Long-chain-fatty-acid--CoA ligase Ornithine carbamoyltransferase Fatty acyl-CoA synthetse Amine oxidase (flavincontaining) Aspartate aminotransferase Fatty acyl-CoA dehydrogenase Carnitine Opalmitoyltransferase Hexokinase Enoyl-CoA hydratase Phosphorylase Inorganic pyrophosphatase 3-hydroxyacyl-CoA dehydrogenase Holocytochrome-c synthase Citrate (si)-synthase Acetyl-CoA Cacyltransferase Glycerol-3-phosphate Oacyltransferase Enoyl-CoA hydratase Arginase [Cytochrome c]-lysine Nmethyltransferase Propionyl-CoA carboxylase RNA polymerase Laminaribiose phosphorylase Creatine kinase DNA-directed DNA polymerase Phospholipase D Cholesterol monooxygenase (side-chain cleaving). Malate dehydrogenase Phospholipase C Phospholipase A2 Glycine aminotransferase Lysophospholipase H(+)-transporting ATP synthase Ketoglutarate dehydrogenase complex De mitochondriële ademhalingsketen defecten de elektronentransportketen defecten de oxidatieve fosforylering De elektronen transportketen.( de ademhalingsketen). Bij de ademhaling wordt zuurstof opgenomen dat via de longen en de bloedbaan getransporteerd wordt naar de mitochondriën in de cel. De functie van zuurstof is de verschillende brandstofmoleculen te oxideren, waarbij energie vrijkomt die voor tal van processen benut kan worden. Bij deze oxidatiereacties worden uiteindelijk elektronen van een substraat overgedragen op zuurstof. In biologische oxidaties vindt deze elektronenoverdracht echter niet rechtstreeks plaats, maar verloopt via een vaste serie reacties die bekend staat als de elektronen transportketen of de ademhalingsketen. Aan de elektronen transportketen neemt een aantal verbindingen deel die ieder voor zich goed een reversibele redoxreactie kunnen ondergaan. Iedere schakel in de keten reduceert een volgende schakel en wordt daarbij zelf geoxideerd. Het NAD+/NADH-redoxkoppel heeft de meest negatieve reductie potentiaal en kan dus het gemakkelijkst elektronen doorgeven aan andere systemen. Het elektronenverloop in de elektronen transportketen volgt de thermodynamisch strikt logische route van NADH ( twee elektronenoverdracht ) via de flavinen FMN of FAD naar coënzym Q. Dit coënzym draagt vervolgens de elektronen via vijf cytochromen ( b,c1,c,a en a3; één-elektronprocessen ) over op zuurstof (vier-elektronenproces). Coënzym Q is een knooppunt in de elektronen transportketen, want het sluist de elektronen van twee verschillende redoxpaden door naar zuurstof via het cytochromensysteem. Coënzym Q is de enige verbinding in de elektronen transportketen die niet sterk gebonden zit aan een eiwit. De isopreenketen aan coënzym Q maakt de verbinding zeer apolair, waardoor deze zich gemakkelijk in de apolaire omgeving van de mitochondriën kan bewegen en zodoende een mobiele schakel vormt tussen flavoproteïnen en cytochromen. Bij het elektronentransport vindt een geleidelijke verlaging van energie plaats. Een deel van de energie wordt opgeslagen in de vorm van ATP doordat het elektronentransport in de mitochondriën gekoppeld is aan de productie van ATP uit ADP en fosfaat.( ATP-adenosine tri-fosfaat;ADP-adenosine di-fosfaat ). Dit proces wordt oxidatieve fosforylering genoemd. Reacties: NAD+ + H+ + 2 e NADH riboflavine + 2H+ + 2 e dihydroriboflavine coënzym Q10 + 2H+ + 2 e dihydrocoënzym Q10 cytochroom c (Fe3+)+ e cytochroom c (Fe2+) cytochroom a3 (Fe3+) + e cytochroom a3 (Fe2+) O2 + 4H+ + 4 e 2H2O De oxidatieve fosforylering ( oxidative phosphorylation or OXPHOS). Oxidatieve fosforylering : Oxidatie is het proces waarbij elektronen worden afgestaan en fosforylering is het proces waarbij een fosfaatgroep wordt opgenomen. Oxidatie vindt plaats in de volgende omkeerbare reactie : NADH + ubiquinone <=> NAD+ + ubiquinol Fosforylering vindt plaats in de volgende omkeerbare reactie : ATP + H2O <=> ADP + fosfaat De oxidatieve fosforylering is verantwoordelijk voor de meeste ATP (adenosine tri-fosfaat) die de cellen nodig hebben. ATP is de bron van cel-energie in alle levende wezens en niet alleen noodzakelijk om te leven, maar voor praktisch alle lichaamsfuncties, door de elektrische energie die daarvoor nodig is. Het OXPHOS pad omvat meer dan 100 polypeptiden wiens genen of in het DNA van de celkern– of in het DNA van het mitochondrion gelokaliseerd zijn. De expressie van deze genen en de samenstelling van de vijf OXPHOS enzym complexen ( I t/m V) is een zeer geordend en gecoördineerd proces. Oxphos bestaat uit vijf enzym complexen. Deze enzymen zijn gelokaliseerd in het mitochondriële binnenste membraan en worden als volgt benoemd: Complex I : NADH: ubiquinone oxidoreductase ( EC 1.6.5.3 ) Complex II : succinate:ubiquinone oxidoreductase ( EC 1.3.5.1 ) Complex III : ubiquinol: ferrocytochroom c oxidoreductase ( EC 1.10.2.2 ) Complex IV: ferrocytochroom c : oxygen oxidoreductase of cytochroom c oxidase ( EC 1.9.3.1 ) Compex V : H(+)-transporting two-sector ATPase ( EC 3.6.3.14 ) Formerly EC 3.6.1.34 Complex I en II collecteren de elektronen uit verschillende bronnen en geven ze af aan ubiquinone (coënzym Q10). De elektronen bewegen zich dan door de complexen III en IV en reageren dan uiteindelijk met zuurstof. Het is nu duidelijk dat ziekten kunnen ontstaan door mutaties in de genen van de celkern of in genen van het mitochondrion en er kunnen zelfs ziekten ontstaan door de fouten in systemen die de interactie tussen deze twee coördineren. Om deze redenen hebben OXPHOS ziekten een complexe reeks van erfelijkheid en hebben een breed spectrum aan klinische presentaties. De enzym complexen in de binnenste mitochondriële membraan: Complex I, III en IV overspannen de binnenste mitochondriële membraan en zijn protonen pompen. Complex II bevindt zich tussen complex I ( NADH: ubiquinone oxidoreductase ) en Complex III ( ubiquinol: ferrocytochroom c oxidoreductase ). Complex II overspant de binnenste mitochondriële membraan niet, maar bevindt zich aan de matrix-zijde van de binnenste mitochondriële membraan en is het ingangspunt van elektronen afkomstig van FADH 2. Complex I is het ingangspunt voor elektronen afkomstig van NADH. Alle 4 genoemde enzymen bevatten prosthetische groepen die de eigenlijke elektronendragers in de enzymen zijn. In elk complex en ook van complex naar complex zijn verschillende groepen betrokken bij de eigenlijke elektronen overdracht. Deze groepen kunnen worden onderscheiden in: ijzer-zwavel proteïnen; haem; koper en flavinen. Allen dienen als elektronendrager, echter elk enzymcomplex wordt geassocieerd met bepaalde prostethetische groepen: Complex I: FMN en Fe-S Complex II: FAD en Fe-S Complex III: Heme b-562, Heme b-566, Heme c1, Fe-S Complex IV: Heme a, Heme a3, CuA, CuB Animatie van Elektronen transport in Mitochondriën Copyright 1997. Thomas M. Terry, The University of Connecticut. Het schema hierboven illustreert een mitochondrion. Bekijk in de animatie hoe NADH H + ionen en elektronen verplaatst in de elektronen transportketen. 1. Protonen worden over het membraan getransporteerd; dat wil zeggen: vanuit de matrix naar de ruimte tussen de binnenste en de buitenste membraan. 2. Elektronen worden in het membraan getransporteerd. 3. Zuurstof is de uiteindelijke elektronenacceptor. Gecombineerd met elektronen en H + ionen wordt water geproduceerd. 4. Terwijl NADH steeds meer H+ en elektronen in de elektronen transportketen overbrengt, zal de proton gradiënt toenemen, met een opbouw van H+ buiten de binnenste mitochondriële membraan en OH- binnen de binnenste mitochondriele membraan. Animatie van ATP synthese in Mitochondriën. Stap 1: Een Proton gradiënt wordt opgebouwd doordat NADH elektronen in de elektrontransportketen brengt.. + Stap 2: Protonen (aangeduid door lading) komen terug in de mitochondriële matrix door kanalen in het ATP synthase enzym complex. ATP wordt gesynthetiseerd uit ADP en fosfaat (Pi) Bekijk in bovenstaande animatie hoe er een opbouw plaatsvindt van H + ionen in de ruimte tussen de binnenste en buitenste mitochondriele membraam telkens wanneer NADH geproduceerd wordt uit oxidatieve reacties. protonen komen de matrix weer binnen door middel van het ATP synthase complex waarna ATP geproduceerd wordt. Protonen worden over het membraan getransporteerd; dat wil zeggen: vanuit de matrix naar de ruimte tussen de binnenste en de buitenste membraan. Terwijl NADH steeds meer H+ en elektronen in de elektronen transportketen overbrengt, zal de proton gradiënt toenemen, met een opbouw van H+ buiten de binnenste mitochondriële membraan en OH- binnen de binnenste mitochondriele membraan. Er ontstaat een proton gradiënt. ATP synthase is een groot proteïne complex met een proton kanaal die de herintreding van protonen toestaat. ATP synthese wordt aangedreven door de resulterende stroom van protonen die door het membraan stromen: ADP + Pi ---> ATP Animaties: ATP synthase http://www.stolaf.edu/people/giannini/flashanimat/metabolism/atpsyn1.swf This short animation illustrates the production of ATP by ATP synthase. ATP synthase mechanism http://www.stolaf.edu/people/giannini/flashanimat/metabolism/atpsyn2.swf This quick animation provides a look at the action of ATP synthase. Mitochondrial electron transport http://www.stolaf.edu/people/giannini/flashanimat/metabolism/mido%20e%20transport.swf Click here for a brief but clear animation demonstrating the electron transport chain. De erfelijkheid. Algemeen. De genen zijn een deel van de chromosomen die op hun beurt gelokaliseerd zijn in de nucleus van elke cel. De chromosomen zijn opgebouwd uit een verbinding die DNA wordt genoemd. De mitochondriën, die zich buiten de nucleus in het cytoplasma van de cel bevinden, zijn een andere plaats waar DNA wordt gevonden. Dus ook de mitochondriën bevatten genen; alhoewel het mitochondriële DNA één lange streng genen is; en niet is samengesteld uit chromosomen. De hoeveelheden mitochondriën in elke cel kan variëren van 1 tot duizenden. Al deze mitochondriën; en dus ook het DNA in de mitochondriën, stammen af van een klein aantal mitochondriën die aanwezig waren in de originele eicel op het moment van de conceptie. Het sperma draagt niet bij aan de mitochondriën van de baby. Dus de mitochondriën worden alleen geërfd van de moeder. (Zeer zeldzaam voorkomend worden mitochondriën van de vader geërfd ). Een abnormaliteit in één van de genen in de mitochondriën kan daardoor worden doorgegeven in de eicellen van de moeder. Daar de meeste eicellen van de moeder dezelfde mitochondriële mutatie bevatten, is het risico op een volgend kind met een mitochondriëleaandoening hoog. Dit patroon van erfelijkheid wordt maternale overerving genoemd. (maternaal = van de moeder). Veel aangeboren mitochondriële aandoeningen worden nucleus defecten genoemd (Een fout in een gen in de kern van de cel); met een overerving van: ofde vader, of de moeder of van zowel de vader als de moeder. Dit patroon wordt autosomaalrecessief erfelijk genoemd. Deze kunnen dominant of recessief zijn. In dit geval is de kans op een aandoening in het volgend kind 25%. Verder is er nog een X-gebonden overerving; deze heeft betrekking op defecten van het X-chromosoom. Nog een andere mogelijkheid is de kans dat er tijdens de bevruchting een mutatie is opgetreden in een gen en dat dit kind de eerste in een familie is die ziek wordt. De effecten van mutaties in de mitochondriële genen. De eicel bevat veel mitochondriën en elk mitochondrion bevat verschillende kopieën van de mitochondriële genen. Wanneer een specifiek mitochondrieel gen in elk mitochondrion in de eicel gemuteerd zou zijn en daarom de verkeerde boodschap uit zou zenden,dan zou de breuk in de energie productie zo ernstig zijn dat de vroege embryo dit niet zou overleven. ( Anders gezegd: pas wanneer een aanvankelijk laag percentage gemuteerde mtDNAs na een zeker aantal celdelingen een bepaalde drempel overschrijdt, leidt dit (soms vrij plotseling) tot problemen. Omdat het aantal mtDNA kopieën per mitochondrion en per cel, alsmede de behoefte aan oxidatieve fosforylering per weefsel verschilt, heeft elk weefsel zijn eigen typische drempelwaarde. ) Dus; een persoon die geboren is met een erfelijke mitochondriële ziekte moet twee typen mitochondriën van zijn of haar moeder hebben geërfd: enkele die de correcte kopieën van het gen bevatten en enkele die de gemuteerde en ziekmakende kopieën van het gen bevatten. In tegenstelling tot de meeste genetische ziekten waarbij de mutatie in alle cellen van het lichaam wordt gezien, worden mutaties van het mitochondriële DNA niet in alle cellen van het lichaam gezien. Mitochondriën worden willekeurig verdeeld in de eicel wanneer deze gevormd wordt in het ovarium. Daarom zal de samenstelling van de mitochondriën in elke eicel variëren van meest normaal tot meest abnormaal. Daarom zullen alle kinderen van deze moeder enige abnormale genen erven, maar alleen symptomen ontwikkelen wanneer de mitochondriën met het gemuteerde gen een zeker kritisch niveau hebben bereikt met de interferentie van de energie productie in het lichaam. Het is dus niet te voorspellen in welke mate iemand ziek zal worden en welke organen daar eventueelbetrokken bij zijn. Evenmin is het te voorspellen wat het verloop van de ziekte zal zijn over een bepaalde tijd, daar men weinig zal kunnen zeggen over de verdeling en de hoeveelheid van de mitochondriën met het gemuteerde gen in het lichaam. De gemuteerde mitochondriën kunnen gelokaliseerd zijn in de skeletspieren, of in het hart ,of de nieren, of in het brein of in de lever, enzovoort. Of in combinaties bij bovenstaande , of in alle organen. Dit maakt de mitochondriële aandoeningen zo complex. Mitochondriële ziekten: Tot de mitochondriële ziekten behoren o.a. : De enzym stoornissen van de citroenzuurcyclus ; Ziekten van het Pyruvaat Dehydrogenase Complex en Pyruvaat Carboxylase ; De Mitochondriële Vetzuur-Oxidatie Stoornissen en defecten van de bèta oxidatie spiraal. Ziekten van de oxidatieve fosforylering. ( indeling naar complexen) of ziekten van de oxidatieve fosforylering ( indeling naar syndromen ). Lactaat Acidemie ( lactic acidemia ): Ziekten van Pyruvaat Dehydrogenase en Pyruvaat Carboxylase 1. Pyruvate Dehydrogenase Complex, E1-Alpha polypeptide 1; PDHA1 Het pyruvaat dehydrogenase complex bestaat uit meerdere kopieën van 3 enzymen : pyruvate decarboxylase (E1; EC 1.2.4.1 ), dihydrolipoyl transacetylase (E2; EC 2.3.1.12 ), dihydrolipoyl dehydrogenase (E3; EC 1.8.1.4 ). e-Medicine: Pyruvate Dehydrogenase Complex Deficiency OMIM: Pyruvate Dehydrogenase Deficiency ; 312170 OMIM: Clinical Synopsis 2. Pyruvate Decarboxylase Deficiency Synoniemen: Intermittent Ataxia with Pyruvate Dehydrogenase or Pyruvate Decarboxylase Deficiency; Ataxia with Lactic Acidose I; PDH Deficiency OMIM: 312170 OMIM: Clinical Synopsis Expasy: Pyruvate decarboxylase EC 4.1.1.1 Enzym Synoniemen: Alpha-carboxylase; Pyruvic decarboxylase; Alpha-ketoacid carboxylase. A 2-oxo acid <=> an aldehyde + CO2 3. Pyruvate Dehydrogenase Phosphatase Deficiency ExPASy: [Pyruvate dehydrogenase (lipoamide)]-phosphatase. EC 3.1.3.43 4. Mitochondrial Phosphoenolpyruvate Carboxykinase 2; PCK 2 ( PEPCK 2 ) OMIM: Phosphoenolpyruvate Carboxykinase Deficiency; PEPCK Deficiency; OMIM 261650 OMIM: Clinical Synopsis ExPASy: Phosphoenolpyruvate carboxykinase (GTP). EC 4.1.1.32 Enzym Synoniemen: Phosphopyruvate carboxylase; Phosphoenolpyruvate carboxylase; Phosphoenolpyruvate;carboxykinase; PEP carboxykinase; PEPCK. GTP + oxaloacetate <=> GDP + phosphoenolpyruvate + CO 2 5. Lactic Acidosis due to Lipoamide Dehydrogenase Deficiency Synoniemen: LAD Deficiency; Congenital Infantile Lactic Acidosis due to LAD Deficiency;Dihydrolipoamide dehydrogenase deficiency; DHLD Deficiency; DLD Deficiency;Maple Sirup Urine Disease Type III; MSUD Type III. OMIM: 238331 OMIM: Clinical Synopsis ExPASy: Dihydrolipoamide dehydrogenase EC 1.8.1.4 Enzym Synoniemen: Lipoamide reductase (NADH); E3 component of alpha-ketoacid dehydrogenase complexes; Lipoyl dehydrogenase; Dihydrolipoyl dehydrogenase; Diaphorase. Dihydrolipoamide + NAD+ <=> lipoamide + NADH 6. Pyruvate Carboxilase Deficiency. Synoniemen: PC Deficiency; Ataxia with lactic acidosis II; Leigh Syndrome due to Pyruvate Carboxylase Def.; Leigh Necrotizing Encephalopathy due to pyruvate carboxylase deficiency OMIM: 266150 OMIM: Clinical Synopsis e-Medicine: Pyruvate Carboxylase Deficiency ExPASy: Pyruvate carboxylase EC 6.4.1.1 Enzym Synoniemen: Pyruvic carboxylase ATP + pyruvate + HCO3- <=> ADP + phosphate + oxaloacetate Mitochondriële Vetzuur-oxidatie stoornissen en defecten van de bèta-oxidatie spiraal: Stoornissen in de Carnitine cyclus: 1. Carnitine Palmitoyltransferase I Deficiency Synoniemen: Hypoketotic hypoglycemie with deficiency of carnitine palmitoyltransferase I; CPT I Deficiency; Hepatic CPT deficiency type I. OMIM: 255120 OMIM: Clinical Synopsis Extra informatie: Carnitine palmitoyltransferase stoornissen Extra informatie: The Spiral Notebook ExPASy: Carnitine O-palmitoyltransferase EC 2.3.1.21 Palmitoyl-CoA + L-carnitine <=> CoA + L-palmitoylcarnitine 2. Carnitine Palmitoyltransferase II Deficiency ( Adult onset ) Synoniemen: Myopathy with deficiency of carnitine palmitoyltransferase II; Myopathic CPT II deficiency. OMIM: 255110 OMIM: Clinical Synopsis Extra informatie: Carnitine palmitoyltransferase stoornissen Extra informatie: The Spiral Notebook ExPASy: Carnitine O-palmitoyltransferase EC 2.3.1.21 Palmitoyl-CoA + L-carnitine <=> CoA + L-palmitoylcarnitine 3. Carnitine Palmitoyltransferase II Deficiency ( Infantile ) Synoniemen: CPT II deficiency, hepatic; OMIM: 600649 OMIM: Clinical Synopsis Extra informatie: Carnitine palmitoyltransferase stoornissen Extra informatie: The Spiral Notebook ExPASy: Carnitine O-palmitoyltransferase EC 2.3.1.21 Palmitoyl-CoA + L-carnitine <=> CoA + L-palmitoylcarnitine 4. Carnitine Palmitoyltransferase II Deficiency ( Lethal neonatal ) OMIM: 608836 OMIM: Clinical Synopsis Extra informatie: Carnitine palmitoyltransferase stoornissen Extra informatie: The Spiral Notebook ExPASy: Carnitine O-palmitoyltransferase EC 2.3.1.21 Palmitoyl-CoA + L-carnitine <=> CoA + L-palmitoylcarnitine 5. Systemic Primary Carnitine Deficiency Synoniemen: Systemic carnitine deficiency due to defect in renal reabsorption of carnitine; Primary carnitine deficiency; Deficiency of carnitine transporter, plasma membraneCarnitine uptake defect; CUD OMIM: 212140 OMIM: Clinical Synopsis e-Medicine: Carnitine deficiency 6. Carnitine/Acylcarnitine Translocase Deficiency Synoniemen: CACT deficiency OMIM: 212138 OMIM: Clinical Synopsis 7. Myopathic Carnitine Deficiency OMIM: 212160 OMIM: Clinical Synopsis Extra informatie: e-Medicine: Carnitine deficiency 8. 3-Hydroxy-3-methylglutaryl-CoA lyase deficiency Synoniemen: HMG-CoA Lyase deficiency; HL Deficiency; Hydroxymethylglutaricaciduria OMIM: 246450 OMIM: Clinical Synopsis ExPASy: Hydroxymethylglutaryl-CoA lyase EC 4.1.3.4 Enzym synoniemen: 3-hydroxy-3-methylglutarate-CoA lyase; HMG-CoA lyase (S)-3-hydroxy-3-methylglutaryl-CoA <=> acetyl-CoA + acetoacetate defecten van de bèta-oxidatie spiraal: 1. 2,4-Dienoyl-CoA Reductase Deficiency OMIM: 222745 OMIM: Clinical Synopsis ExPASy: 2,4-dienoyl-CoA reductase (NADPH). EC 1.3.1.34 Enzym synoniemen: 4-enoyl-CoA reductase (NADPH). Trans-2,3-didehydroacyl-CoA + NADP+ <=> trans,trans-2,3,4,5-tetradehydroacyl-CoA + NADPH 2. Short Chain Acyl Co-A Dehydrogenase deficiëntie; SCAD Synoniemen: ACADS Deficiency; SCADH Deficiency; SCAD Deficiency; Lipid Storage Myopathy secondary to Short Chain Acyl-CoA Dehydrogenase Deficiency. OMIM: 201470 OMIM: Clinical Synopsis OMIM: Short Chain Acyl-CoA Dehydrogenase; SCAD; Acyl-CoA Dehydrogenase, C2 to C3 short chain Extra informatie : Korte Keten Acyl-CoA Dehydrogenase Deficiëntie ExPASy: Butyryl-CoA dehydrogenase EC 1.3.99.2 Enzym synoniemen: Short-chain acyl-CoA dehydrogenase; Unsaturated acyl-CoA reductase; Butyryl dehydrogenase Butanoyl-CoA + ETF <=> 2-butenoyl-CoA + reduced ETF 3. 3-α-Hydroxyacyl-CoA Dehydrogenase; HADH ( voorheen: Short Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency; SCHAD) OMIM: 601609 ExPASy: 3-hydroxyacyl-CoA dehydrogenase EC 1.1.1.35 Enzyn synoniemen: Beta-hydroxyacyl dehydrogenase; Beta-keto-reductase (S)-3-hydroxyacyl-CoA + NAD+ <=> 3-oxoacyl-CoA + NADH 4. Medium Chain Acyl Co-A Dehydrogenase deficiency; MCAD Synoniemen: ACADM Deficiency; MCADH Deficiency; Carnitine deficiency secondary to medium-chain Acyl-CoA Dehydrogenase deficiency. OMIM: 210450 OMIM: Clinical Synopsis OMIM: Medium-Chain Acyl-CoA Dehydrogenase e-Medicine: Medium-Chain Acyl-CoA Dehydrogenase Deficiency GeneReviews: Medium-chain acyl-coenzyme A dehydrogenase (MCAD) deficiency Centers for Disease Control and Prevention: MCADD MCAD: Rijks Universiteit Groningen: Factsheet MCAD MCAD: MCADAngel ExPASy: Acyl-CoA dehydrogenase EC 1.3.99.3 Enzym synoniemen: Medium-chain acyl-CoA dehydrogenase; Acyl dehydrogenas Acyl-CoA + ETF <=> 2,3-dehydroacyl-CoA + reduced ETF Long Chain Acyl Co-A Dehydrogenase deficiëntie; LCAD Synoniemen: ACADL deficiency; Nonketotic Hypoglycemia due to deficiency of Long Chain Acyl-CoA Dehydrogenase OMIM: 201460 OMIM: Clinical Synopsis e-Medicine: Long-Chain Acyl CoA Dehydrogenase Deficiency ExPASy: Long-chain acyl-CoA dehydrogenase EC 1.3.99.13 Acyl-CoA + ETF <=> 2,3-dehydroacyl-CoA + reduced ETF Hydroxyacyl-CoA Dehydrogenase / 3-Ketoacyl-CoA Thiolase / Enoyl-CoA Hydratase; Alpha subunit; HADHA Synoniemen: Trifunctional protein, alpha subunit; Mitochondrial Trifunctional protein, Alpha subunit; Long Chain Hydroxyacyl-CoA Dehydrogenase; LCHAD OMIM: 600890 Trifunctional Protein Deficiency, Type I, Included; Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency, Included. OMIM: Clinical Synopsis The Fatty Oxidation Disorders (FOD) Family Support Group™ : LCHAD e-Medicine: LCHAD deficiency, trifunctional protein deficiency Hydroxyacyl-CoA Dehydrogenase / 3-Ketoacyl-CoA Thiolase / Enoyl-CoA Hydratase; Beta subunit; HADHB Synoniemen: Trifunctional Protein, Beta SubunitHydroxyacyl-CoA Dehydrogenase; HADH OMIM: 143450 Trifunctional Protein Deficiency, Type II, included; Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency, included; LCHAD Deficiency, included; 3-Hydroxydicarboxylicaciduria, included; Combined enzyme defect of mitochondrial fatty acid oxidation, included. OMIM: Clinical Synopsis The Fatty Oxidation Disorders (FOD) Family Support Group™ : LCHAD e-Medicine: LCHAD deficiency, trifunctional protein deficiency ExPASy: Long-chain-3-hydroxyacyl-CoA dehydrogenase EC 1.1.1.211 (S)-3-hydroxyacyl-CoA + NAD+ <=> 3-oxoacyl-CoA + NADH Very Long Chain Acyl-CoA Dehydrogenase Deficiency; VLCAD OMIM: 210475 OMIM: Clinical Synopsis Glutaric Aciduria Type IIA ( Glutaar Acidurie Type IIA ) Ethylmalonic-Adipicaciduria ( EMA ); Multiple Acyl-CoA Dehydrogenase Deficiency ( MADD ); Deficiency of Elektron Transfer Flavoprotein, Alpha polypeptide; Deficiency Synoniemen: GA IIA; Glutaricacidemia IIA; of ETFA. OMIM: 231680 OMIM: Clinical Synopsis Extra informatie: Glutaar acidurie type II Glutaric Aciduria Type IIB ( Glutaar Acidurie Type IIB ) Synoniemen: Elektron Transfer Flavoprotein, beta polypeptide; GA IIB. OMIM: 130410 Extra informatie: Glutaar acidurie type II Glutaric Aciduria Type IIC ( Glutaar Acidurie Type IIC ) Synoniemen: Electron transfer flavoprotein: ubiquinone oxidoreductase (ETF:QO) deficiency; GA IIC; Glutaricacidemia IIC; Electron transfer flavoprotein: Dehydrogenase Deficiency. OMIM: 23167 OMIM: Clinical Synopsis ExPASy: Electron-transferring-flavoprotein dehydrogenase EC 1.5.5.1 Enzym synoniemen: Electron transfer flavoprotein-ubiquinone oxidoreductase; ETF-QO; ETF-ubiquinone oxidoreductase; ETF dehydrogenase Reduced ETF + ubiquinone = ETF + ubiquinol Extra informatie: Glutaar acidurie type II Mitochondrial Porin Mitochondriële Porine VDAC: Voltage Dependent Anion Channel Voltage Afhankelijk Anion Kanaal Meer informatie: VDAC Web Page Meer informatie: Structure of the voltage dependent, ion selective channel (VDAC) 1. Voltage-Dependent Anion Channel 1; VDAC1 OMIM: Voltage Dependent Anion Channel Deficiency ( VDAC Deficiency ) 2. Voltage-Dependent Anion Channel 2; VDAC2 OMIM: Voltage-Dependent Anion Channel 2; VDAC2 Stoornissen van de Ketonlichamen stofwisseling: 1. Mitochondrial HMG-CoA synthase deficiency OMIM: 600234 2. 3-hydroxy-3-methylglutaryl CoA lyase deficiency Synoniemen: HMG-CoA Lyase deficiency; HL deficiency; Hydroxymethylglutaric aciduria OMIM: 246450 OMIM: Clinical Synopsis ExPASy: EC 4.1.3.4 Hydroxymethylglutaryl-CoA lyase Synoniemen: 3-hydroxy-3-methylglutarate-CoA lyase; HMG-CoA lyase 3. R-3-hydroxybutyrate deficiency 4. Succinyl-CoA:3-oxoacid CoA transferase deficiency ( SCOT deficiency ) Synoniemen: Ketoacidosis due to SCOT deficiency OMIM: 245050 OMIM: Clinical Synopsis 5. Alpha-methylacetoaceticaciduria ( Mitochondrial acetoacetyl-CoA thiolase deficiency ) Synoniemen: ; T2 deficiency; MAT deficiency; Beta ketothiolase deficiency; 2-methyl-3-hydroxybutyric acidemia; 3-oxothiolase deficiency; OMIM: 203750 OMIM: Clinical Synopsis Ziekten van de Oxidatieve Fosforylering De ademhalingsketen defecten: ( indeling van de ziektes naar de verschillende complexen ) 1. Deficiency of Mitochondrial Respiratory Chain Complex I ( Mitochondriële Ademhalingsketen Complex I deficiëntie ) Synoniemen: NADH: Q ( I ) Oxidoreductase Deficiency; Mitochondrial Complex I Deficiency; Deficiency of Mitochondrial NADH Dehydrogenase Component of Complex I; Mitohondrial Myopathy with deficiency of respiratory chain NADH-CoQ Reductase activity. OMIM: 252010 OMIM: Clinical Synopsis Extra Informatie: Complex I Homepage ExPASy: NADH dehydrogenase (ubiquinone). EC 1.6.5.3 Enzym synoniemen: Ubiquinone reductase; Type 1 dehydrogenase; Complex 1 dehydrogenase; Coenzyme Q reductase; Complex I (electron transport chain); Complex I (mitochondrial electron transport); Complex I (NADH:Q1 oxidoreductase); Dihydronicotinamide adenine dinucleotide-coenzyme Q reductase; DPNH-coenzyme Q reductase; DPNH-ubiquinone reductase; Mitochondrial electron transport complex 1; Mitochondrial electron transport complex I; NADH coenzyme Q1 reductase; NADH-coenzyme Q oxidoreductase. NADH-coenzyme Q reductase; NADH-CoQ oxidoreductase; NADH-CoQ reductase. NADH-ubiquinone reductase; NADH-ubiquinone oxidoreductase; NADH-ubiquinone-1 reductase. Reduced nicotinamide adenine dinucleotide-coenzyme Q reductase; NADH:ubiquinone oxidoreductase complex. NADH-Q6 oxidoreductase; Electron transfer complex I NADH + ubiquinone <=> NAD+ + ubiquinol 2. Deficiency of Mitochondrial Respiratory Chain Complex II ( Mitochondriële Ademhalingsketen Complex II deficiëntie ) Synoniemen: Succinate-CoQ reductase deficiëntie OMIM: 252011 OMIM: Clinical Synopsis ExPASy: Succinate dehydrogenase (ubiquinone). EC 1.3.5.1 Enzym Synoniemen: Succinic dehydrogenase Succinate + ubiquinone <=> fumarate + ubiquinol 3. Deficiency of Mitochondrial Respiratory Chain Complex III ( Mitochondriële Ademhalingsketen Complex III deficiëntie ) Synoniemen: Cytochrome related disease of muscle and nervous system OMIM: 124000 OMIM: Clinical Synopsis ExPASy: Ubiquinol--cytochrome c reductase EC 1.10.2.2 Enzym synoniemen: Ubiquinone-cytochrome c oxidoreductase; Cytochrome bc1 complex.; Complex III (mitochondrial electron transport). QH2 + 2 ferricytochrome c <=> Q + 2 ferrocytochrome c 4. Deficiency of Mitochondrial Respiratory Chain Complex IV ( Mitochondriële Ademhalingsketen Complex IV deficiëntie ) Synoniemen: Cytochroom c oxidase deficiency ; COX deficiency OMIM: 220110 OMIM: Clinical Synopsis Extra Informatie: Cytochroom Oxidase Homepage NORD: cytochrome c oxidase deficiency ExPASy: Cytochrome-c oxidase. EC 1.9.3.1 Enzym synoniemen: Cytochrome oxidase; Cytochrome a(3); Cytochrome aa(3);Warburg's respiratory enzyme; Complex IV (mitochondrial electron transport). 4 ferrocytochrome c + O2 <=> 4 ferrocytochrome c + H2O 5. ATPase deficiency. ( Mitochondriële Ademhalingsketen Complex V deficiëntie ) Synoniemen: ATP Synthase Deficiency OMIM: 604273 ExPASy: H(+)-transporting two-sector ATPase. EC 3.6.3.14 Formerly EC 3.6.1.34 Enzym synoniemen: ATP synthase; F(1)-ATPase; F(0)F(1)-ATPase; H(+)-transporting ATP synthase;H(+)transporting ATPase; Mitochondrial ATPase; Chloroplast ATPase. ATP + H2O + H+ (In) <=> ADP + phosphate + H+ (Out) Ziekten van de Oxidatieve Fosforylering: De ademhalingsketen defecten: ( Indeling van de ziektes als verschillende syndromen ) 1. Alpers progressive sclerosing poliodystrophy OMIM: 203700 OMIM: Clinical Synopsis NINDS: Alpers' disease Who named it?: Bernard Jacob Alpers 2. Barth syndrome Synoniemen: Cardioskeletal Myopathy with Neutropenia and abnormal mitochondria, MGA Type II; 3-α-Methylglutaconicaciduria Type II OMIM: 302060 OMIM: Clinical Synopsis Extra Informatie: Barth Syndroom 3. Friedreich ataxia (FRDA). OMIM: 229300 OMIM: Clinical Synopsis GeneReviews: Friedreich ataxia e-Medicine: Friedreich ataxia Who named it?: Nikolaus Friedreich 4. Hereditary spastic paraplegia (HSP). e-Medicine: Hereditary Spastic Paraplegia GeneReviews: Hereditary Spastic Paraplegia 5. Kearns-Sayre syndrome (KSS). Synoniemen: Ophthalmoplegia, pigmentary degeneration of the retina, and cardiomyopathy; Oculocraniosomatic syndrome; Ophtalmoplegia-Plus syndrome; Mitochondrial Cytopathy OMIM: 530000 OMIM: Clinical Synopsis e-Medicine: Kearns - Sayre Syndrome ( KSS ) NINDS: Kearns-Sayre Who named it?: Thomas P. Kearns ; George Pomeroy Sayre 6. Leber hereditary optic neuroretinopathy (LHON). OMIM: 535000 OMIM: Clinical Synopsis GeneReviews: Leber Hereditary Optic Neuropathy Who named it?: Theodor Karl Gustav von Leber 7. Leigh subacute necrotizing encephalomyopathy Synoniemen: Leigh Syndrome; LS OMIM: 256000 OMIM: Clinical Synopsis Extra informatie: Ziekte van Leigh GeneReviews: Mitochondrial DNA-associated (mtDNA-associated) Leigh syndrome and NARP 8. Mitochondrial encephalomyopathy with lactic acidosis and strokelike episodes (MELAS). OMIM: 540000 OMIM: Clinical Synopsis e-Medicine: MELAS Syndrome GeneReviews: MELAS MedicineNet: MELAS 9. Mitochondrial myopathy, peripheral neuropathy, gastrointestinal, and encephalopathy disease (MNGIE). OMIM: 603041 10. Mohr-Tranebjaerg syndrome (DFN1). Synoniemen: Dystonia-Deafness Syndrome; DDS; Deafness-Dystonia-Optic atrophy syndrome; DDP OMIM: 304700 OMIM: Clinical Synopsis OMIM: 300356 TIMM8A GeneReviews: Deafness and Hereditary Hearing Loss Overview GeneReviews: Mohr-Tranebjaerg syndrome 11. Myoclonic epilepsy with ragged red fibers (MERRF). OMIM: 545000 OMIM: Clinical Synopsis GeneReviews: MERRF Extra informatie: MERRF 12. Neurogenic muscle weakness, ataxia, retinitis pigmentosa (NARP). OMIM: 551500 OMIM: Clinical Synopsis GeneReviews: Mitochondrial DNA-associated (mtDNA-associated) Leigh syndrome and NARP 13. Pearson syndrome Synoniemen: Sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction OMIM: 557000 OMIM: Clinical Synopsis e-Medicine: Pearson Syndrome 14. Progressive external ophthalmoplegia (PEO). OMIM: 157640 OMIM: Clinical Synopsis 15. Syndromic forms of sensorineural hearing loss 16. Wolfram syndrome Synoniemen: ► Diabetes insipidus and mellitus with optic atrophy and deafness; ► DIDMOAD Syndrome OMIM: 598500 OMIM: Clinical Synopsis Extra informatie: Worldwide Society of Wolfram Syndrome Families