K.U.Leuven
Gevorderde
Anorganische Chemie
Koen Binnemans
Leuven
2007
Inhoudstafel
Hoofdstuk 1:
Oxidaties en reducties ................................................................................. 4
1.1 Basisconcepten ................................................................................................................ 4
1.2 Electrochemische cellen .................................................................................................. 7
1.2.1 Galvanische en elektrolytische cellen....................................................................... 7
1.2.2 Elektromotieve kracht .............................................................................................11
1.2.3 Elektrodepotentialen................................................................................................13
1.2.4 Nernst-vergelijking..................................................................................................16
1.2.5 Standaardelektrodepotentiaal...................................................................................17
1.3 Overpotentiaal ................................................................................................................19
1.4 Redoxstabiliteit in water.................................................................................................20
1.4.1 Reacties met water...................................................................................................20
1.4.2 Disproportionering en comproportionering.............................................................22
1.4.3 Oxidatie door luchtzuurstof .....................................................................................23
1.5 Schematische voorstelling van redox-eigenschappen ....................................................24
1.5.1 Latimer-diagrammen ...............................................................................................24
1.5.2 Frost-diagrammen....................................................................................................28
1.5.3 Pourbaix-diagrammen .............................................................................................33
1.6 Corrosie van ijzer ...........................................................................................................39
1.7 Metallurgische processen ...............................................................................................43
1.7.1 Ellingham-diagrammen ...........................................................................................43
1.7.2 Pyrometallurgie .......................................................................................................47
1.7.3 Productie van ijzer ...................................................................................................51
1.7.4 Elektrometallurgie: Productie van aluminium.........................................................56
Hoofdstuk 2:
Metaalclusters .............................................................................................59
2.1 Isolobale moleculaire fragmenten ..................................................................................59
2.2 Metaal-metaalbindingen .................................................................................................65
2.3 Meervoudige metaal-metaalbindingen ...........................................................................66
2.4 Polymetallische clusters .................................................................................................70
2.5 Zintl-ionen ......................................................................................................................78
Hoofdstuk 3:
Karakteristieke structuren van vaste stoffen ...........................................80
3.1 Moleculaire kristallen .....................................................................................................80
3.2 Bolstapelingen ................................................................................................................81
3.3 Metalen en legeringen ....................................................................................................87
3.3.1 Metaalbinding..........................................................................................................87
3.3.2 Structuur van metalen in de vaste toestand .............................................................88
1
3.3.3 Polymorfie bij metalen ............................................................................................89
3.3.4 Legeringen...............................................................................................................91
3.4 Structuren van covalente kristallen.................................................................................92
3.5 Structuren van ionische verbindingen ............................................................................96
3.5.1 Straalverhoudingen en coördinatiepolyeders...........................................................96
3.5.2 Structuren van binaire ionische verbindingen .......................................................100
3.5.3 Structuur van ternaire ionische verbindingen ........................................................106
3.6 Structuren van verbindingen met complexe ionen .......................................................110
3.7 Silicaatstructuren ..........................................................................................................113
3.8 Chemische formules van anorganische kristalverbindingen ........................................116
Hoofstuk 4:
Niet-stoechiometrische verbindingen.....................................................118
4.1 Thermodynamica en onvolmaakte kristallen................................................................118
4.2 Typen van roosterfouten...............................................................................................119
4.3 Niet-stoechiometrische verbindingen ...........................................................................126
4.4 Vaste oplossingen.........................................................................................................127
4.4.1 Substitutionele vaste oplossingen..........................................................................128
4.4.2 Interstitiële vaste oplossingen................................................................................131
4.5 Glas...............................................................................................................................132
4.5.1 Chemische samenstelling en structuur van glas ....................................................132
4.5.2 Glaseigenschappen ................................................................................................138
4.5.3 Glasbereiding.........................................................................................................141
4.5.4 Glasbewerking.......................................................................................................143
Hoofdstuk 5:
Interpretatie van fasediagrammen..........................................................146
5.1 Faseregel, fasen, componenten, vrijheidsgraden en evenwicht ....................................146
5.2 Unaire systemen ...........................................................................................................150
5.2.2 Fasediagram van zwavel........................................................................................154
5.2.2 Fasediagram van water ..........................................................................................156
5.2.3 Fasediagram van siliciumdioxide ..........................................................................158
5.3 Binaire systemen ..........................................................................................................159
5.3.1 Eenvoudige binaire systemen met een eutecticum ................................................161
5.3.2 Binaire systemen met verbindingen ......................................................................164
5.3.3 Binaire systemen met vaste oplossingen ...............................................................167
5.3.4 Binaire systemen met vast-vast fasetransities........................................................174
5.4 Ternaire systemen.........................................................................................................175
Hoofdstuk 6:
Synthesemethoden in de vaste-stofchemie..............................................180
6.1 Hoge-temperatuursmethoden (keramische methoden) .................................................180
6.2 Verbrandingssynthese...................................................................................................186
2
6.3 Hydrothermale synthese ...............................................................................................189
Bronvermelding ....................................................................................................................192
1
2
H
He
3
4
5
6
7
8
9
10
Li
Be
B
C
N
O
F
Ne
11
12
13
14
15
16
17
Al
Si
P
S
C l Ar
31
32
33
34
Na Mg
19
K
37
20
38
Rb Sr
55
56
39
40
41
42
Y
Z r Nb Mo T c R u R h P d Ag C d In
57
72
C s Ba La
Hf
88
104
87
Fr
30
24
C r Mn F e C o Ni
89
R a Ac R f
58
73
74
Ta W
105
106
43
75
44
76
27
29
23
V
25
26
28
22
Ti
21
Ca Sc
45
77
46
78
C u Z n G a G e As S e
47
79
48
80
49
81
R e Os
Ir
P t Au Hg T l
107
109
110
108
111
50
51
52
S n S b Te
82
83
Pb Bi
84
35
18
36
Br Kr
53
I
85
54
Xe
86
P o At R n
112
D b S g B h Hs Mt D s R g Uub
59
60
61
62
63
64
65
66
67
68
69
70
71
C e P r Nd P m S m E u G d T b D y Ho E r T m Y b L u
90
91
Th Pa
92
U
93
94
95
96
97
98
99
100
101
102
103
Np P u Am C m B k C f E s F m Md No L r
3
Hoofdstuk 1:
Oxidaties en reducties
1.1 Basisconcepten
Een redoxreactie houdt de transfer van elektronen van een bepaald deeltje naar een ander
in. Een deeltje wordt geoxideerd als het elektronen verliest. Het wordt gereduceerd wanneer
het elektronen opneemt. Bijgevolg is oxidatie een verlies van elektronen en reductie is een
opname van elektronen. Een oxiderend reagens (of oxidans) neemt elektronen op van een
andere verbinding en wordt gereduceerd. Een reducerend reagens (of reductans) geeft
elektronen aan een andere verbinding en wordt tijdens dit proces geoxideerd. In de reactie:
Fe3+ + V2+ → Fe2+ + V3+
is Fe3+ het oxidans omdat het een elektron van V2+ opneemt. V2+ is het reductans omdat het
een elektron aan Fe3+ geeft. Fe3+ wordt gereduceerd and V2+ wordt geoxideerd als de reactie
van links naar rechts verloopt.
Redoxreacties lijken in vele opzichten op zuur/base-reacties. In beide gevallen
worden één of meer geladen deeltjes van een donor naar een acceptor getransfereerd. De
deeltjes zijn elektronen in redoxreacties en protonen in zuur/base-reacties. Herinner dat
volgens de Brønsted-Lowry-theorie een zuur na afgave van een proton in de (geconjugeerde)
base wordt omgezet. Een base wordt door opname van een proton in het geconjugeerde zuur
omgezet. Op een analoge wijze zijn de producten van een redoxreactie een geconjugeerd
oxidans en een geconjugeerd reductans (alhoewel de term “geconjugeerd” zelden of nooit
voor redoxreacties wordt gebruikt). Beschouw het volgende redox-evenwicht:
ox1 + red2 ' red1 + ox2
Het oxidans ox1 accepteert elektronen van reductans red2 ter vorming van aan nieuw
reductans red1. Tegelijkertijd wordt red2 door afgave van elektronen in een oxidant ox2
omgezet. Als het evenwicht van dit proces de producten begunstigt, dan is ox1 een efficiëntere
elektronenacceptor (en dus een sterker oxidans) dan ox2, het deeltje dat ontstaat uit red2 door
afgave van elektronen. Eveneens kan men dan zeggen dat red2 een efficiëntere
elektronendonor (en dus een sterkere reductor) dan red1 is.
Het is vaak nuttig om de redoxreactie in twee halfreacties op te splitsen, waarvan één de
oxidatie beschrijft en de andere de reductie. Beschouw bijvoorbeeld de oxidatie van
ijzer(II)ionen door permanganaat-ionen in zuur milieu:
4
5Fe2+ + MnO4- + 8H+ ' 5 Fe3+ + Mn2+ + 4H2O
De overeenkomstige halfreacties zijn:
MnO4- + 8 H+ + 5e ' Mn2+ + 4 H2O
5Fe2+ ' 5Fe3+ + 5e
Deze vergelijkingen tonen duidelijk aan dat het permanganaation de elektronenacceptor is en
bijgevolg het oxidans, terwijl het ijzer(II)-ion de elektronendonor en reductans is. Merk ook
op dat in feite de tweede halfreactie kan door deling door 5 vereenvoudigd worden. Als men
twee halfreacties combineert, moeten het aantal elektronen in de twee halfreacties echter
gelijk zijn.
Wanneer de elektronen van een redoxreactie doorheen een elektrische stroomkring lopen,
dan kunnen we iets over de reactie leren door de elektrische stroom en de
potentiaalverschillen in de stroomkring te meten. De elektrische stroom in de stroomkring is
immers recht evenredig met de snelheid van de redoxreactie (of elektrochemische reactie),
terwijl de celspanning recht evenredig is met de verandering van vrije energie van de
redoxreactie. De elektrische lading (q) wordt gemeten in coulomb (C). De grootte van de
lading van één elektron bedraagt 1,602 × 10-19 C, zodat één mol elektronen een lading van
9,6485 × 104 C (= 96485 C) heeft. Deze lading wordt de Faraday-constante (F) genoemd. Het
verband tussen de lading q en het aantal mol n is:
q=n⋅F
(1.1)
De hoeveelheid lading die iedere seconde door het circuit stroomt wordt de
elektrische stroom (I) genoemd. De eenheid van elektrische stroom is de ampère (A). Een
stroom van één ampère komt overeen met een lading van één coulomb per seconde die langs
een punt in het circuit stroomt.
Het potentiaalverschil (E) tussen twee punten is een maat voor de arbeid die nodig is (of
kan geleverd worden) wanneer een elektrische lading zich van één plaats naar de andere
verplaatst. Potentiaalverschillen worden in volt (V) uitgedrukt. Arbeid heeft de dimensies van
een energie, en de overeenkomstige eenheid is de joule (J). Wanneer een lading q zich
doorheen een potentiaalverschil E beweegt, wordt de volgende arbeid geleverd:
5
Arbeid = E ⋅ q
(1.2)
Er komt één joule aan energie vrij of er wordt één joule aan energie opgenomen als een lading
van 1 coulomb zich beweegt tussen twee punten waartussen het potentiaalverschil 1 volt
bedraagt. 1 volt is bijgevolg 1 J/C. Hoe groter het potentiaalverschil tussen twee punten is, des
te sterker zal de “druk” op het geladen deeltje zijn om tussen deze twee punten te reizen. Een
batterij van 12 V drukt de elektronen acht maal sterker door een elektrische stroomkring dan
een batterij van 1,5 V.
De verandering van de Gibbs vrije energie, ΔG, voor een chemische reactie die
omkeerbaar wordt uitgevoerd bij een constante temperatuur en druk komt overeen met de
maximaal mogelijke elektrische arbeid die kan verricht worden door de reactie op zijn
omgeving:
Arbeid verricht op de omgeving = -ΔG
(1.3)
Het negatief teken in deze vergelijking duidt aan dat de vrije energie van een systeem daalt als
het arbeid op de omgeving verricht. Combinatie van vergelijkingen 1.1, 1.2 en 1.3 levert:
ΔG = - arbeid = - E ⋅ q
(1.4)
De vergelijking die het verschil in vrije energie in verband brengt met het elektrisch
potentiaalverschil is:
ΔG = - n F E
(1.5)
De wet van Ohm zegt dat de elektrische stroom (I) die door een elektrische stroomkring
loopt recht evenredig is met het potentiaalverschil (E) over de stroomkring en omgekeerd
evenredig met de elektrische weerstand (R) van de stroomkring:
I = E/R
(1.6)
De eenheid van weerstand is de ohm (Ω). Een stroom van 1 ampère zal stromen door een
stroomkring met een potentiaalverschil van 1 volt als de weerstand van de stroomkring 1 Ω
bedraagt. Hoe groter het potentiaalverschil is, hoe groter de elektrische stroom zal zijn. Hoe
groter de weerstand is, hoe minder stroom door de stroomkring zal lopen.
6
Het vermogen (P) is de arbeid die per eenheid van tijd wordt verricht. De SI-eenheid van
vermogen is J/s, beter bekend als de watt (W).
P = arbeid/t = (E⋅q)/t = E⋅(q/t)
(1.7)
Aangezien q/t de elektrische stroom I is, kunnen we schrijven:
P = E⋅I
(1.8)
Een cel die in staat is om een stroom te leveren bij een potentiaalverschil van 1 volt levert een
vermogen van 1 watt. De weerstand is de enige plaats in de stroomkring waar energie
verloren gaat. Het vermogen is gelijk aan de snelheid waarmee warmte in de weerstand wordt
geproduceerd.
1.2 Electrochemische cellen
1.2.1 Galvanische en elektrolytische cellen
De meeste redoxreacties kunnen op twee verschillende manieren worden uitgevoerd. Bij
de eerste methode worden het oxidans en het reductans samen gebracht in een milieu waarin
de reagerende moleculen, atomen of ionen dicht genoeg bij elkaar kunnen komen om een
directe elektronentransfer te laten plaats vinden. Bij de tweede methode zijn de reagentia een
deel van een elektrochemische cel, waarin de twee halfreacties fysisch van elkaar gescheiden
zijn. De elektronentransfer vindt hier plaats via een metallische geleider van elektriciteit.
Elektrochemische cellen koppelen bijgevolg redoxreacties aan een externe elektrische
stroomkring. Electrochemische cellen kunnen in twee klassen worden onderverdeeld,
naargelang ze elektrische energie produceren of verbruiken. Als de redoxreactie spontaan
verloopt, gebruikt de elektrochemische cel de Gibbs vrije energie die door de reactie
beschikbaar is om chemische energie in elektrische energie om te zetten. De elektrische
energie wordt dan aan de externe stroomkring geleverd waar deze nuttige arbeid kan
verrichten. Een elektrochemische cel die een spontaan verlopende chemische reactie gebruikt
om elektriciteit te genereren wordt een galvanische cel (of voltaïsche cel) genoemd.
Elektrolytische cellen werken omgekeerd: zij onttrekken elektrische energie aan een externe
stroomkring en gebruiken deze om reacties uit te voeren die niet spontaan kunnen verlopen.
In beide typen van elektrochemische cellen wordt de grootte en de richting van de stroom in
7
de externe stroomkring bepaald door de verandering van de Gibbs vrije energie van de
redoxreactie in de cel.
Electrochemische cellen bestaan uit twee elektroden, de ene wordt een anode genoemd en
de andere een kathode. Beide zijn in een elektroliet-oplossing gedompeld. In de meeste cellen
verschillen de samenstelling en de concentratie van de elektroliet-oplossingen, en de interface
tussen beide oplossingen noemt men een vloeibare junctie. Enkele typen van cellen werken
zonder vloeibare junctie. Hierbij zijn de twee elektrolietoplossingen identiek.
De anode van een electrochemische cel is de elektrode waar de oxidatie plaats vindt. De
kathode is de elektrode waar de reductie plaats vindt. Typische kathodische processen zijn:
Ag+ + e ' Ag(s)
2H+ + 2e ' H2(g)
Fe3+ + e ' Fe2+
IO4- + 2H+ + 2e ' IO3- + H2O
De laatste drie reacties vinden plaats op het oppervlak van een inerte geleider, zoals platina,
die als kathode dienst doet. Merk op dat zowel anionen als kationen op een platina-elektrode
reductie kunnen ondergaan. Waterstof is vaak het product dat aan de kathode wordt gevormd
in oplossingen die geen deeltjes bevatten die gemakkelijker dan protonen worden
gereduceerd. Typische anode-reacties zijn:
Zn(s) ' Zn2+ + 2e
2Cl- ' Cl2(g) + 2e
Fe2+ ' Fe3+ + e
2H2O ' O2(g) + 4H+ + 4e
De eerste reactie vereist een zinkelektrode, terwijl de andere op een inerte elektrode plaats
vinden. De laatste reactie treedt gewoonlijk op in oplossingen die geen andere deeltjes
bevatten die gemakkelijker dan water kunnen geoxideerd worden.
Voor de bespreking van een galvanische cel beschouwen we de reactie tussen metallisch
zink en koper(II)ionen:
8
Zn(s) + Cu2+(aq) → Zn2+(aq) + Cu(s)
De reactie is illustratief voor een spontaan redoxproces. Het exacte mechanisme waarop de
elektronentransfer verloopt is niet gekend. We kunnen bovenstaande reactie echter weergeven
als een combinatie van twee halfreacties:
Oxidatie:
Zn(s) → Zn2+(aq) + 2e
Reductie:
2e + Cu2+(aq) → Cu(s)
In een galvanische cel vinden deze twee halfreacties op twee verschillende elektroden plaats,
zodat de elektronentransfer plaats vindt door een externe stroomkring in plaats van direct
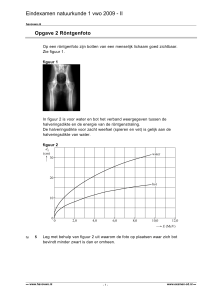
tussen zinkmetaal en koper(II)ionen. De cel die in Figuur 1.1 wordt weergeven is ontworpen
om gebruik te maken van deze reactie om elektriciteit te produceren. De halfcel aan de
linkerkant bevat een koperelektrode en een CuSO4-oplossing. De halfcel aan de rechterkant
bestaat uit een zinkelektrode in een ZnSO4-oplossing. De halfcellen zijn gescheiden door een
poreuze scheidingswand of een zoutbrug die de mechanische menging van de oplossingen
verhindert, maar die wel toelaat dat ionen onder de invloed van een elektrische stroom
passeren. Een galvanische cel van dit type wordt een Daniell-cel (of Daniell-element)
genoemd, ter ere van de Britse chemicus John Frederic Daniell (1790-1845).
Figuur 1.1. Daniell-cel.
9
Wanneer de zink- en koperelektrode door middel van een metalen draad met elkaar
verbonden worden, vloeien elektronen van de zink- naar de koperelektrode. Aan de
zinkelektrode wordt het zinkmetaal tot zink(II)ionen geoxideerd. Deze elektrode is de anode
en de elektronen die het resultaat van de oxidatie zijn verlaten hier de cel. De elektronen
reizen door de externe stroomkring naar de koperelektrode, waar ze voor de reductie van
koper(II)ionen tot metallisch koper gebruikt worden. Het gevormde koper wordt op de
koperelektrode afgezet. De koperelektrode is de kathode. Hier komen de elektronen de
galvanische cel binnen en vindt de reductie plaats. Aangezien elektronen aan de zinkelektrode
worden gevormd, duidt men deze anode als de negatieve pool aan. Bij een galvanische cel in
werking vloeien de elektronen in het externe circuit van de negatieve naar de positieve pool.
De kathode waar elektronen worden gebruikt voor de elektrodereactie, is bijgevolg de
positieve pool. De beweging van ionen in de cel maakt de elektrische stroomkring volledig.
Op het eerste gezicht is het vreemd dat anionen, die negatief geladen zijn, naar de anode, die
de negatieve pool is, migreren. Eveneens is het vreemd dat kationen, die positief geladen zijn,
naar de kathode die de positieve pool is, migreren. Een nauwkeurige beschouwing van de
elektrodereacties geeft echter een oplossing voor dit probleem. Bij de anode worden
zink(II)ionen gevormd en blijven elektronen in het metaal achter. De elektrische neutraliteit
van de oplossing blijft steeds behouden. In de oplossing die de elektrode omgeeft moet er
evenveel negatieve lading van anionen aanwezig zijn als er positieve lading van kationen
aanwezig is. Bijgevolg migreren sulfaat-ionen naar de anode om het effect te neutralizeren
van de zink(II)ionen die daar worden gevormd. Tegelijkertijd migreren zink(II)ionen van de
anode naar de kathode. Bij de kathode worden elektronen gebruikt om koper(II) ionen tot
kopermetaal te reduceren. Terwijl de koper(II)ionen ontladen worden, migreren meer
koper(II)ionen naar de omgeving van de kathode om de plaats in te nemen van de ionen die
aan de oplossing onttrokken zijn. Als dit niet zou gebeuren, zou er zich een overmaat aan
sulfaat-ionen rond de kathode opstapelen.
Een poreuze scheidingswand wordt toegevoegd om te vermijden dat de oplossingen van
de twee halfcellen mechanisch zouden gemengd worden. De scheidingswand is een
keramische schijf of een speciaal membraan. Als de koper(II)ionen met de zinkelektrode in
contact zouden komen, zouden de elektronen direct worden getransfereerd in plaats van
transfer via de externe stroomkring. Bij een normale werking van de galvanische cel treedt
deze kortsluiting niet op omdat de koper(II)ionen migreren in een tegenovergestelde richting
van de zinkelektrode. In plaats van een poreuze scheidingswand wordt vaak een zoutbrug
gebruikt. Dit is een omgekeerde U-vormige buis die een oplossing bevat van een zout zoals
NaNO3, KNO3 of KCl. De uiteinden van de zoutbrug zijn afgesloten met een poreuze plug
bijv. glaswol) of een poreuze glasschijf, die vermijdt dat de twee oplossingen met elkaar
10
worden vermengd, maar die wel de passage van ionen toelaat. Vaak is het elektroliet in de
zoutbrug in de vorm van een gel (bijv. een agar-gel) aanwezig. Bij gebruik van een zoutbrug
kunnen de twee halfcellen zich in twee gescheiden containers bevinden (zoals getoond in
Figuur 1.1). Wanneer de galvanische cel in werking is en de zoutbrug bevat KNO3, dan
migreren K+-ionen van de zoutbrug in het kathode-compartiment en een kleine hoeveelheid
anionen migreert van de kathode in de zoutbrug. De migratie van ionen uit de zoutbrug is
groter dan de migratie van ionen in de zoutbrug, omdat de zoutconcentratie in de zoutbrug
veel hoger is dan de concentratie in de halfcellen. Aan de andere kant van de zoutbrug
migreren NO3-ionen in het anodecompartiment en een kleine hoeveelheden kationen migreert
in de zoutbrug, om een opstapeling van lading te vermijden.
1.2.2 Elektromotieve kracht
Als 1 M oplossingen van ZnSO4 en CuSO4 in de Daniell-cel worden gebruikt, dan kan
deze cel door de volgende notatie worden weergegeven:
Zn(s)⏐Zn2+(1 M)⏐Cu2+(1 M)⏐Cu(s)
Hierbij stellen de verticale lijnen fasegrenzen voor. De conventie is dat de verbinding die de
anode vormt als eerste links wordt geschreven. De andere materialen van de cel worden dan
weergegeven in de volgorde die men tegenkomt als men van de anode naar de kathode in de
cel gaat. De samenstelling van de kathode wordt als laatste weergegeven.
De elektrische stroom die door de galvanische cel wordt geproduceerd is het resultaat van
de elektromotieve kracht (emf) van de cel, die in volt wordt uitgedrukt. De emf van de cel is
des te groter naarmate de neiging van de celreactie om te verlopen groter wordt. De emf van
de cel is echter ook afhankelijk van de concentraties van de verbindingen die gebruikt worden
om de cel te maken en van de temperatuur.
De standaard-emf, E°, heeft betrekking op de elektromotieve kracht van een cel, waarbij
alle reagentia en producten in hun standaardtoestand aanwezig zijn. De E°-waarden worden
gewoonlijk gegeven voor metingen die bij 25 °C werden uitgevoerd. De standaardtoestand
van een vaste stof of een vloeistof zijn natuurlijk de vaste stof en de vloeistof zelf. De
standaardtoestand van een gas of van een verbinding in oplossing wordt gedefinieerd als een
toestand met ideale activiteit 1. Er worden correcties gemaakt om afwijkingen van het ideale
geval door intermoleculaire en interionische interacties in rekening te brengen. We maken de
veronderstelling dat de activiteit van ionen kan weergegeven worden door hun molaire
concentraties en de activiteit van gassen door hun drukken in atmosfeer. Volgens deze
benadering bevat een standaardcel ionen bij een concentratie van 1 mol/L en eventueel gassen
11
bij drukken van 1 atmosfeer. In notaties van elektrochemische cellen worden de concentraties
gewoonlijk enkel weergegeven als deze van de standaardwaarden afwijken.
Als de emf-waarde gebruikt moet worden als een betrouwbare maat voor de neiging van
de celreactie om op te treden, moet het potentiaalverschil de maximale waarde zijn die voor
de beschouwde cel kan bereikt worden. Als er echter een aanzienlijke elektrische stroom door
de stroomkring vloeit tijdens de meting, zal het gemeten potentiaalverschil E kleiner zijn
omwille van de inwendige weerstand van de cel. Als de cel elektrische stroom levert,
veroorzaken
de
elektrodenreacties
bovendien
concentratieveranderingen
die
het
potentiaalverschil doen afnemen. De emf van een cel moet bijgevolg gemeten worden onder
omstandigheden waarbij er omzeggens geen elektrische stroom door de cel vloeit. Deze
meting wordt uitgevoerd door van een potentiometer gebruik te maken. De stroomkring van
een potentiometer bevat een stroombron met een variabel voltage en een opstelling om dit
potentiaalverschil te meten. De elektrochemische cel die bestudeerd wordt, wordt met de
stroomkring van de potentiometer verbonden om zulke manier dat de emf van de cel door de
emf van de potentiometer tegengewerkt wordt. Als de emf van de cel groter is dan deze van
de potentiometer, vloeien de elektronen in de normale richting die ook gevolgd wordt als de
cel spontaan ontladen wordt. Als echter de emf van de stroombron van de potentiometer
groter is dan deze van de cel, vloeien de elektronen in tegengestelde richting en wordt de
celreactie omgekeerd. Wanneer de twee emf-waarden in balans zijn, vloeien er geen
elektronen. Dit potentiaalverschil is de reversibele emf-waarde van de cel. De emf-waarde
van een standaard Daniell-cel is 1,10 V.
De wetten van Faraday zijn zowel op galvanische als op elektrolytische cellen toepasbaar.
Men moet er echter rekening mee houden dat de elektriciteit wordt gegeneerd door de
gelijktijdige oxidatie- en reductiehalfreacties die aan respectievelijk de anode en kathode
plaatsvinden. Beide reacties moeten optreden opdat de cel elektriciteit zou leveren. Twee
faraday aan elektriciteit wordt er gevormd door de oxidatie van 1 mol zink aan de anode
samen met de reductie van 1 mol koper(II)ionen aan de kathode. Als de halfreacties
Anode:
Zn(s) → Zn2+(aq) + 2e
Kathode:
2e + Cu2+(aq) → Cu(s)
gelezen worden in termen van aantal mol, stellen deze de stroom voor van tweemaal het getal
van Avogadro aan elektronen of de vorming van twee faraday aan elektriciteit. De
hoeveelheid elektrische energie (in joule) die door de galvanische cel wordt geleverd (of de
hoeveelheid arbeid die kan verricht worden) is volgens vergelijking 1.2 gelijk aan het product
12
van de hoeveelheid geleverde lading (in coulomb) en de emf van de cel (in volt). De
elektrische energie die geproduceerd wordt door de reactie tussen 1 mol zinkmetaal en 1 mol
koper(II)ionen kan als volgt worden berekend:
2 × 96485 C × 1,10 V = 212267 J ≈ 212 kJ
Merk op dat één volt⋅coulomb gelijk is aan één joule. De emf gebruikt in deze berekening is
de reversibele emf (E°) van de standaard Daniell-cel en bijgevolg het maximale
potentiaalverschil van deze cel. Bijgevolg is 212 kJ de maximale hoeveelheid arbeid die kan
verricht worden door de werking van dit type van cel. De maximale netto arbeid die kan
verkregen worden uit een chemische reactie uitgevoerd bij een constante temperatuur en druk
is een maat voor de afname van de Gibbs vrije energie van het systeem (zie ook vergelijking
1.5). Voor de Daniell-cel is ΔG gelijk dus aan –212 kJ. De verandering in vrije energie die
afgeleid wordt van de E°-waarde, wordt weergeven door het symbool ΔG°. De verandering
van vrije energie van de reactie is een maat voor de neiging van de reactie om op te treden.
Als er op het systeem arbeid moet verricht worden om de verandering te doen plaats vinden,
is de verandering niet spontaan. Bij constante temperatuur en druk is een spontane
verandering een verandering waarbij netto arbeid kan verkregen worden. Bijgevolg zal voor
elke spontane reactie de vrije energie van het systeem dalen: ΔG is negatief. Uit vergelijking
1.5 volgt dat enkel als de E°-waarde positief is, de celreactie spontaan verloopt en als
energiebron kan gebruikt worden.
1.2.3 Elektrodepotentialen
Op dezelfde manier als een celreactie kan beschouwd worden als de som van twee
halfreacties, kan de emf van een cel beschouwd worden als de som van twee halfpotentialen.
Het is echter onmogelijk om de absolute waarde van de potentiaal van een enkelvoudige
halfcel te bepalen. Daarom dient er een relatieve schaal opgesteld te worden, waarbij er de
waarde nul wordt toegekend aan een standaard referentiehalfcel en de halfcelpotentialen
relatief ten opzichte van de referentie-elektrode worden uitgedrukt.
De gebruikte referentiehalfcel is de standaard-waterstofelektrode (SHE). Deze bestaat uit
waterstofgas bij een druk van 1 atmosfeer, dat borrelt over een platina-elektrode (bedekt met
fijn verdeeld platinametaal om de oppervlakte te vergroten) die gedompeld is in een zure
oplossing met een activiteit aan H+-ionen van 1. Door het borrelen van waterstofgas door de
oplossing wordt de oplossing met dit gas verzadigd. Een standaard-waterstofelektrode die
13
door een zoutbrug verbonden is met een standaard Cu2+/Cu elektrode, kan als volgt worden
voorgesteld:
Pt⏐H2⏐H+⏐⏐Cu2+⏐Cu
Een dubbele verticale streep stelt een zoutbrug voor. De waterstofelektrode is de anode, de
koperelektrode is de kathode en de emf van de cel is 0,34 V.
De emf van de cel kan beschouwd worden als de som van de halfcelpotentiaal voor de
oxidatiehalfreactie (die we aanduiden met het symbool Eox°) en de halfcelpotentiaal voor de
reductiehalfreactie (die we aanduiden met het symbool Ered°). Voor de hierboven vermelde cel
hebben we:
Anode:
H2 → 2H+ + 2 e
Eox° = 0,00 V
Kathode:
2e + Cu2+ → Cu
Ered° = + 0,34 V
Aangezien arbitrair de potentiaal 0,00 V aan de standaard-waterstofelektrode wordt
toegekend, kan de emf van de gehele cel toegeschreven worden aan de standaard Cu2+/Cuelektrode. De waarde +0,34 V wordt de standaardelektrodepotentiaal van de Cu2+/Cuelektrode genoemd. Merk op dat de elektrodepotentialen voor reductiehalfreacties worden
gegeven. Als het symbool E° (zonder een subscript) wordt gebruikt, veronderstelt men dat het
over de elektrodepotentiaal Ered° gaat.
Als een cel wordt samengesteld uitgaande van een standaard-waterstofelektrode en een
standaard Zn2+/Zn-elektrode, dan is de zinkelektrode de anode en de emf van de cel is 0,76 V
(Figuur 1.2). Bijgevolg:
Anode:
Zn → Zn2+ + e
Eox° = + 0,76 V
Kathode:
2 H+ + 2e→ H2
Ered° = 0,00 V
The waarde +0,76 V wordt soms de oxidatiepotentiaal genoemd, omdat dat deze overeenkomt
met een oxidatiehalfreactie. Een elektrodepotentiaal is echter een reductiepotentiaal. Om de
elektrodepotentiaal van het Zn2+/Zn-koppel te verkrijgen, moeten we het teken van de
oxidatiepotentiaal omdraaien zodat de potentiaal overeenkomt met de omgekeerde halfreactie,
zijnde een reductie:
2e + Zn2+ → Zn
Ered° = -0,76 V
14
Figuur 1.2 Standaard-waterstofelektrode en een Zn2+/Zn elektrode
Het is niet noodzakelijk om een cel die de standaard-waterstofelektrode bevat te
gebruiken
om
een
standaardelektrodepotentiaal
te
verkrijgen.
Bijvoorbeeld,
de
2+
standaardpotentiaal van de Ni /Ni-elektrode kan verkregen worden uitgaande van de cel:
Ni⏐Ni2+⏐⏐Cu2+⏐Cu
The emf van deze cel is 0,59 V, en de nikkelelektrode treedt als anode op:
Ni + Cu2+ → Ni2+ + Cu
Ecell° = +0,59 V
De standaardelektrodepotentiaal van het Cu2+/Cu-koppel werd voorafgaand bepaald:
2e + Cu2+ → Cu
Ered° = + 0,34 V
Als we de Cu2+/Cu-halfreactie van de celreactie aftrekken en we trekken de halfcelpotentiaal
af van de emf van de cel, vinden we:
Ni → Ni2+ + 2e
Eox° = +0,25 V
15
De elektrodepotentiaal is bijgevolg:
2e + Ni2+ → Ni
Ered° = - 0,25 V
De waarde van de elektrodepotentiaal is afhankelijk van de temperatuur en van de
concentratie van de verbindingen die gebruikt werden om de halfcel samen te stellen. Deze
variabelen worden constant gehouden voor de standaardelektrodepotentialen.
In het algemeen is de potentiaal van een elektrochemische cel, of het nu een galvanische
of een elektrolytische cel is, gelijk aan het verschil tussen de elektrodepotentiaal van de
kathode en de elektrodepotentiaal van de anode:
Ecel = Ekathode – Eanode
(1.9)
Men dient te benadrukken dat zowel Ekathode als Eanode elektrodepotentialen zijn en dat het
bijgevolg de potentialen zijn van halfreacties die als reducties geschreven zijn. Een positief
teken voor Ecel duidt aan dat de redoxreactie spontaan verloopt en dat het over een
galvanische cel gaat. Voor een elektrolytische cel is Ecel negatief. Als een verkorte
voorstelling van een elektrochemische cel wordt geschreven, dan staat bij conventie het
anodisch proces steeds links.
1.2.4 Nernst-vergelijking
De grootte van de elektrodepotentiaal is een maat voor de kracht ten opzichte van de
standaard-waterstofelektrode waarmee een halfreactie naar evenwicht wordt gedreven. Hieruit
volgt dat de numerieke waarden van de elektrodepotentialen concentratie-afhankelijk zijn. Zo
zullen koper(II)ionen in een geconcentreerde koper(II)oplossing gemakkelijker tot koper
worden gereduceerd dan koper(II)ionen in een verdunde oplossing. Bijgevolg is de
elektrodepotentiaal voor koper groter in een geconcentreerdere oplossing.
Een kwantitatief verband tussen de elektrodepotentiaal en concentratie werd in 1889
uitgewerkt door de Duitse chemicus Walther Hermann Nernst (1864-1941). Beschouw de
veralgemeende reversibele halfreactie:
aA + bB + .... + ne ' cC + dD + ….
16
Hierbij staan de hoofdletters voor deeltjes (geladen of ongeladen) die aan de
elektronentransfer deelnemen, e is het elektron, en a, b, c, d en n zijn het aantal mol van de
deelnemende deeltjes in de halfcelreactie. Men kan zowel theoretisch als experimenteel
aantonen dat de elektrodepotentiaal voor dit proces kan beschreven worden door de
vergelijking:
RT [C] [D] ⋅ ⋅ ⋅
ln
nF [A ]a [B]b ⋅ ⋅ ⋅
c
E = Eo −
d
(1.10)
Hierbij is:
Eo = de standaardelektrodepotentiaal, die karakteristiek is voor elke halfreactie;
R = de universele gasconstante, 8,314 J K-1 mol-1;
T = absolute temperatuur (in Kelvin);
n = aantal mol elektronen die verschijnen in de halfreactie van het elektrodeproces;
F = de faraday = 96485 C;
ln = de natuurlijke logaritme = 2,303 log.
Substitutie van de numerieke waarden voor de verschillende constanten in vergelijking 1.10,
omzetting van de natuurlijke logaritme naar de logaritme met grondgetal 10, en specificatie
van 25 °C als de temperatuur, geeft de bekende Nernst-vergelijking:
0,005892
[C] [D] ⋅ ⋅ ⋅
log a b
n
[A] [B] ⋅ ⋅ ⋅
c
E = Eo −
d
(1.11)
De letters tussen de vierkante haken stellen strikt gesproken activiteiten voor, maar meestal
worden voor berekeningen de activiteiten door molaire concentraties vervangen. Als A een
opgeloste stof is, stelt [A] bijgevolg de concentratie van A in mol per liter voor. Als A een gas
is, wordt [A] in vergelijking 1.10 vervangen door pA, zijnde de partiële druk van A in
atmosfeer. Als A voorkomt als een zuivere vloeistof in overmaat als een tweede fase, als een
zuivere vaste stof in overmaat als een tweede fase, of als A het solvent water is, dan nemen
we aan dat [A] de waarde 1 heeft.
1.2.5 Standaardelektrodepotentiaal
Alhoewel de standaardelektrodepotentiaal E° reeds in sectie 1.2.3 aan bod is gekomen,
willen we er hier toch nog een aantal bedenkingen op een rij zetten. Uit vergelijking (1.11)
17
volgt dat de elektrodepotentiaal van de halfcel gelijk is aan de standaardelektrodepotentiaal
als de logaritmische term nul is. Dit is het geval als de activiteiten van alle reagentia en
producten gelijk aan 1 zijn. De standaardelektrodepotentiaal is een belangrijke fysische
constante die een kwantitatieve beschrijving geeft van de relatieve drijfkracht naar evenwcht
voor een halfcelreactie.
De standaardelektrodepotentiaal is een relatieve grootheid in de zin dat het de potentiaal
is van een electrochemische cel waarin de anode bestaat uit de standaardwaterstofelektrode,
waaraan arbitrair een potentiaalwaarde van nul werd toegekend (bij om het even welke
temperatuur). Bijgevolg refereren de standaardelektrodepotentialen enkel naar halfcelreacties
die als reducties geschreven zijn. De standaardelektrodepotentialen zijn relatieve
reductiepotentialen.
De standaardelektrodepotentiaal is een maat voor de relatieve grootte van de drijfkracht
voor een halfreactie. Zijn numerieke waarde is onafhankelijk van de notatie die gebruikt
wordt om de halfreactie in uit te drukken. Als we het het linker- en rechterlid van een
halfreactie met een factor vermenigvuldigen, zal de waarde van E° niet veranderen:
Ag+ + e ' Ag(s)
E°= +0,799 V
5Ag+ + 5e ' 5Ag(s)
E°= +0,799 V
Het teken van de standaardelektrodepotentiaal geeft een aanwijzing voor de neiging van
het betrokken deeltje om gereduceerd te worden, relatief ten opzichte van de neiging van
protonen om gereduceerd te worden. Een positief teken duidt aan dat het oxidans (met alle
reagentia en producten aanwezig met een activiteit 1) gemakkelijker wordt gereduceerd dan
protonen en dat de halfcel zich als een kathode gedraagt wanneer deze met de
standaardwaterstofelektrode wordt gekoppeld. Een negatief teken toont aan het betrokken
deeltje een zwakker oxidans dan protonen is en dat de elektrode zich gedraagt als een anode
in een galvanische cel die de standaardwaterstofelektrode bevat.
Naast de standaardelektrodepotentiaal gebruikt men soms ook formele potentialen om te
compenseren voor activiteitseffecten en voor fouten die te wijten zijn aan het bestaan van
evenwichten die met elkaar in competitie staan. De formele potentiaal van een systeem is de
potentiaal van een halfcel (ten opzichte van de standaardwaterstofelektrode) wanneer de
concentratie van elke opgeloste stof die aan de halfreactie deelneemt gelijk is aan 1 mol/L en
de concentraties van alle andere opgeloste stoffen nauwkeurig worden gespecifieerd. Met
andere woorden, de formele potentiaal is de reductiepotentiaal die geldig is in het geval van
een set van gespecifieerde voorwaarden (pH, ionische sterkte, concentratie van
18
complexvormende agentia, ...). Er kunnen grote verschillen optreden tussen een formele
potentiaal en de standaardelektrodepotentiaal. Bijvoorbeeld, de standaardelektrodepotentiaal
voor de reductie van ijzer(III) naar ijzer(II) is 0,771V. In een 1M oplossing van perchloorzuur
is de formele potentiaal voor dezelfde halfreactie 0,731 V. Het verschil is te wijten aan het feit
dat de activiteitscoëfficiënt van ijzer(III) aanzienlijk kleiner is dan deze van ijzer(II) bij de
grote ionische sterkte van een 1M HClO4 oplossing. Bijgevolg is de verhouding van de
activiteiten van beide deeltjes kleiner dan 1, wat volgens de Nernst-vergelijking leidt tot een
kleinere waarde voor de elektrodepotentiaal. In een 1M HCl oplossing bedraagt de formele
potentiaal van dit koppel slechts 0,700 V. In dit geval is de activiteitsverhouding
ijzer(III)/ijzer(II) zelfs nog kleiner omdat de chlorocomplexen van ijzer(III) stabieler zijn dan
deze van ijzer(II). Dit resulteert in een grotere afwijking van de standaardelektrodepotentiaal.
In de biochemie wordt er met formele potentialen gewerkt in plaats van met
standaardelektrodepotentialen. De standaardelektrodepotentiaal zijn geldig voor een pHwaarde van 0 ([H+] = 1 mol/L of beter a(H+) = 1). Omdat de pH in dierlijke of plantaardige
cellen in de buurt van 7 is, zijn elektrodepotentialen die van toepassing zijn op pH = 0 niet erg
geschikt. De formele potentiaal bij pH 7 wordt door biochemici E°’ genoemd.
1.3 Overpotentiaal
De thermodynamische gegevens die uit de standaardelektrodepotentiaal kunnen afgeleid
worden laten toe om te bepalen of een reactie spontaan kan verlopen onder de heersende
omstandigheden. De thermodynamische gegevens zeggen iets over de neiging van deze
reactie om op te treden. Thermodynamica zegt echter niets over de snelheid waarmee deze
spontane reactie plaatsvindt.
Een negatieve waarde voor de standaardelektrodepotentiaal van een metaalion/metaalredoxkoppel duidt aan dat het metaal in staat is om H+-ionen of om het even welk positiever
koppel onder standaardvoorwaarden in waterige oplossing te reduceren. Het is echter niet
zeker of er een mechanistische weg bestaat om deze reductie te verwezenlijken. Er bestaan
geen algemene regels om te voorspellen of een gegeven reactie snel zal verlopen, aangezien
de factoren die de snelheid van redoxreacties beïnvloeden heel complex zijn. Een nuttige
vuistregel is dat redoxkoppels waarvan de potentiaal 0,6 V lager is dan de potentiaal van het
H+/H2-koppel in staat zijn om bij de heersense pH de reductie van H+-ionen tot H2-gas tegen
een redelijke snelheid uit te voeren. Evenzeer zijn koppels met een potentiaal die 0,6 V hoger
is dan de potentiaal van het O2,H+/H2O koppel bij de heersende pH in staat om water tegen
een redelijke snelheid te oxideren. De overpotentiaal kan gedefinieerd worden als de
19
potentiaal bovenop de evenwichtspotentiaal (situatie zonder stroom) die moet aangelegd
worden opdat de redoxreactie tegen een redelijke snelheid zou verlopen.
Het bestaan van de overpotentiaal verklaart waarom bepaalde metalen in staat zijn om
zuren te reduceren, maar niet water zelf. De metalen (waaronder Fe en Zn) hebben negatieve
reductiepotentialen, maar deze potentialen zijn niet negatief genoeg om de vereiste
overpotentiaal in een neutrale oplossing (pH = 7) te verwezenlijken. Het verschil E(H+,H2) –
E(Fe2+/Fe) kan vergroot worden als E(H+/H2) positiever wordt gemaakt, wat mogelijk is door
de pH te verlagen. Wanneer dit verschil groter wordt dan de overpotentiaal, zal het metaal
reductie van H+-ionen tegen een redelijke snelheid doen plaats vinden. De commerciële
elektrolytische bereiding van zwaar water (D2O) is afhankelijk van de grotere overpotentiaal
voor de vorming van D2 dan voor de vorming van H2.
1.4 Redoxstabiliteit in water
Een ion of molecule in oplossing kan vernietigd worden door oxidatie of door
reductie die veroorzaakt wordt door om het even welk deeltje dat in de oplossing aanwezig is.
Om de stabiliteit van een deeltje in oplossing te bepalen, moet rekening houden met factoren
zoals het solvent, het opgelost deeltje zelf, de andere opgeloste stoffen en opgeloste zuurstof.
Aangezien water het meest gebruikte solvent is om redoxreacties te bestuderen, is het nuttig
om de redoxstabiliteit van opgeloste deeltjes in water nader te bekijken.
1.4.1 Reacties met water
Water kan als oxidans optreden, waarbij het tot H2 wordt gereduceerd:
2H2O(l) + 2 e → H2(g) + 2 OH-(aq)
E° = -0,83 V + (0,059 V)pOH
De potentiaal vereist voor reductie van H3O+ is minder negatief:
2H+(aq) + 2e → H2(g)
E° = -(0,059V)pH
Deze reactie treedt op als men spreekt over de “reductie van water”. Water kan ook als
reductans optreden, waarbij het tot O2 wordt geoxideerd:
O2(g) + 4H+(aq) + 4e → 2H2O(l)
E°= 1,23V-(0,059V)pH
Redoxactieve deeltjes kunnen in water enkel blijven bestaan als hun potentiaal gelegen is
tussen de limieten die door deze processen worden gedefinieerd.
20
De reacties van metalen met water of met een waterig zuur is in feite de oxidatie van het
metaal door water of door protonen, omdat de totale reacties één van de volgende processen
is:
M(s) + H2O → M+(aq) + 1/2H2(g) + OH-(aq)
M(s) + H+ → M+(aq) + 1/2H2(g)
Hierbij wordt enkel de vorming van éénwaardige ionen beschouwd. De reacties voor de
vorming van meerwaardige ionen zijn analoog. Wanneer de E°-waarde negatief is, ondergaat
het metaal oxidatie in een 1 M zuuroplossing ter vorming van H2-gas. De reacties kunnen
echter traag verlopen, zoals blijkt uit de rol van de overpotentiaal die we hogerop hebben
besproken. Alhoewel de reacties van aluminium en magnesium met vochtige lucht traag
verlopen, kunnen de metalen gedurende jaren worden gebruikt in de aanwezigheid van water
en zuurstof. Deze metalen overleven omdat ze gepassiveerd worden, of tegen reactie
beschermd worden door de vorming van een ondoordringbare oxidelaag. Aluminiumoxide en
magnesiumoxide vormen een beschermende huid op de onderliggende metaallaag. Een
gelijkaardige vorm van passivering bestaat er bij koper en zink.
Aangezuurd water is een zwak reductans, behalve ten opzichte van sterk oxidantia zoals
Co3+(aq). De E°-waarde van het redoxkoppel Co3+/Co2+ bedraagt +1,92 V. Co3+(aq) wordt
door water gereduceerd ter vorming van O2:
4Co3+(aq) + 2H2O(l) ' 4Co2+(aq) + O2(g) + 4H+(aq)
E = +0,69 V
Aangezien er bij deze reactie protonen worden gevormd, begunstigt een lagere zuurheid van
de oplossing (hogere pH) de reactie, aangezien verlaging van de protonenconcentratie het
redoxevenwicht naar rechts verschuift. Slechts weinig oxiderende stoffen (Ag2+ is een ander
voorbeeld) zijn in staat om water snel genoeg te oxideren zodat er redelijke snelheiden voor
de O2-vorming worden waargenomen. De redoxkoppels Ce4+/Ce3+ (+1,76 V), Cr2O72-/Cr3+
(+1,38 V) en MnO4-/Mn2+ (+1,51 V) in zuur milieu hebben allen weliswaar een E°-waarde die
groter is dan de +1,23 V die theoretisch vereist is om water te oxideren, maar door de
overpotentiaal zijn deze reacties te traag om te worden waargenomen. De oorzaak van de
hoge activatie-energie bij de oxidatie van water is de vereiste om vier elektronen te
transfereren ter vorming van een zuurstof-zuurstof dubbele binding. Door gebruik te maken
van een geschikte katalysator zijn deze redoxkoppels wel in staat om water oxidatief te
21
ontbinden. Het onderzoek naar efficiënte katalysatoren om water te ontbinden is een uitdaging
voor anorganische chemici.
Een reducerend reagens dat snel water tot H2 kan reduceren, of een oxiderend reagens dat
water snel tot O2 kan oxideren, kan in een waterige oplossing niet overleven. Het
stabiliteitsgebied van water, dat in Figuur 1.3 wordt getoond, is het potentiaal- en pH-gebied
waarbinnen water thermodynamisch stabiel is ten opzichte van oxidatie en reductie. De
getoonde lijnen zijn een grafische voorstelling van de pH-afhankelijkheid van de oxidatie en
reductie van water zoals deze hogerop in deze sectie werden besproken. Redoxkoppels die in
water thermodynamisch onstabiel zijn liggen buiten de grenzen die door het stabiliteitsgebied
worden gedefineerd. Deze koppels bestaan hetzij uit een sterk reductans (beneden de lijn voor
H2-vorming), hetzij uit een sterk oxidans (boven de lijn voor O2-vorming). Deze redoxkoppels
zijn in staat om water tot H2 te reduceren of water tot O2 te oxideren. Het stabiliteitsgebied
voor natuurlijke waters worden in Figuur 1.3 weergegeven voor twee verticale lijnen bij pH =
4 en bij pH = 9. Deze pH-waarden duiden de limieten aan die gewoonlijk in
oppervlaktewaters worden gemeten.
Figuur 1.3. Stabiliteitsgebied van water.
1.4.2 Disproportionering en comproportionering
Aangezien E°(Cu+,Cu) = +0,52 V en E°(Cu2+,Cu+) = +0,16 V, en aangezien beide
potentialen in het stabiliteitsgebied van water liggen, zijn Cu+-ionen niet in staat om water te
oxideren of te reduceren. Desondanks is koper(I) niet stabiel in water, omdat het
disproportionering kan ondergaan. Een disproportionering is een redoxreactie waarbij het
22
oxidatiegetal van een element tegelijkertijd wordt verhoogd en verlaagd. Een element dat
disproportionering ondergaat treedt op als zijn eigen oxiderend en reducerend reagens. De
reactie:
2Cu+(aq) → Cu2+(aq) + Cu(s)
is het verschil van de volgende twee halfreacties:
Cu+(aq) + e- → Cu(s)
E° = +0,52 V
Cu2+(aq) + e- → Cu+(aq)
E° = +0,16 V
De disproportionering verloopt spontaan, omdat E = (0,52 V) – (0,16 V) = +0,36 V.
Het
omgekeerde
van
een
disproportionering
is
een
comproportionering.
Comproportionering is een redoxreactie waarbij twee deeltjes van eenzelfde element in een
verschillende oxidatietoestand een product vormen waarbij het element in een intermediaire
oxidatietoestand is. Een voorbeeld is:
Ag2+(aq) + Ag(s) → 2 Ag+(aq)
E° = +1,18 V
Deze grote potentiaalwaarde toont aan dat zilver(II) en zilver(0) in water volledig tot zilver(I)
worden omgezet.
1.4.3 Oxidatie door luchtzuurstof
Wanneer een oplossing aan de lucht wordt blootgesteld, bijvoorbeeld omdat de oplossing
zich in een open bekerglas bevindt, moet men de mogelijkheid van reactie tussen de opgeloste
stoffen en opgelost zuurstofgas in overweging nemen. Beschouw bijvoorbeeld een oplossing
van Fe2+. De standaardredoxpotentiaal van het Fe3+/Fe2+-redoxkoppel bedraagt 0,77 V. Deze
waarde is gelegen in het stabiliteitsgebied van water, wat doet veronderstellen dat Fe2+ in
water blijft bestaan. We kunnen uit de standaardredoxpotentiaalwaarden ook afleiden dat de
oxidatie van metallisch ijzer door H+(aq) slechts tot ijzer(II) verloopt, omdat verdere oxidatie
tot ijzer(III) thermodynamisch onder standaardomstandigheden ongunstig is. De situatie
verandert echter drastisch in de aanwezigheid van O2. Ijzer(III) is inderdaad de meest stabiele
vorm van ijzer in de aardkorst en de meeste ijzer in sedimentgesteenten werd daarin uit
waterig milieu onder de vorm van ijzer(III) afgezet. De reactie:
23
4Fe2+(aq) + O2(g) + 4H+(aq) → 4Fe3+(aq) + 2H2O(l)
is het verschil van de volgende twee halfreacties:
O2(g) + 4H+(aq) + 4e- → 2H2O(l)
E = +1,23 V – (0,059)pH
Fe3+(aq) + e- → Fe2+(aq)
E° = +0,77 V
wat inhoudt dat de E°-waarde voor de oxidatie van Fe2+ door O2 +0,46V bedraagt en dat de
oxidatie van Fe2+ door O2 spontaan verloopt (bij pH = 0). Omdat +0,46 V geen voldoende
grote overpotentiaal is, verloopt de oxidatie van ijzer(II) in waterige oplossing door
luchtzuurstof in de afwezigheid van een katalysator traag. Bijgevolg kan een waterige Fe2+oplossing een tijdlang worden bewaard in het laboratorium zonder dat bijzondere
beschermingsmaatregelen moeten worden genomen.
1.5 Schematische voorstelling van redox-eigenschappen
Er bestaan een aantal nuttige samenvattingen in diagramvorm van de relatieve
thermodynamische stabiliteiten van een serie van deeltjes waarbij één element voorkomt in
een reeks van verschillende oxidatiegetallen.
1.5.1 Latimer-diagrammen
Een Latimer-diagram geeft schematisch de standaardpotentiaal van redoxkoppels
weer en wordt gebruikt om kwantitatieve elektrochemische gegevens in compacte vorm
samen te vatten en maakt relaties tussen deeltjes duidelijk. De numerische waarde van de
standaardredoxpotentiaal E° (in V) wordt geschreven boven een horizontale lijn die deeltjes
van een element in verschillende oxidatietoestanden verbindt. De meest geoxideerde vorm
van het element wordt links geschreven en bij de deeltjes rechts hiervan komt het element in
opeenvolgende lagere oxidatietoestanden voor. Soms worden de oxidatiegetallen onder (of
boven) de deeltjes geschreven. Als typisch voorbeeld kan het Latimer-diagram van chloor in
zuur midden worden gegeven:
24
De notatie:
1.20
ClO 4− ⎯+⎯
⎯→ ClO3−
stelt voor:
ClO4-(aq) + 2H+(aq) + 2e- → ClO3-(aq) + H2O(l)
E0 = +1,20 V
Op analoge wijze is
1, 67
HClO ⎯+⎯
⎯→ Cl 2
een verkorte schrijfwijze voor:
2 HClO(aq) + 2H+(aq) + 2e- → Cl2(g) + 2H2O(l) E0 = +1,67 V
Het omvormen van een Latimer-diagram tot een halfreactie houdt vaak in dat men de
atoombalans in evenwicht moet brengen door het toevoegen van deeltjes die in zuur midden
aanwezig zijn (bijv. H+ en H2O). De elektrische lading wordt dan in evenwicht gebracht door
een geschikt aantal elektronen toe te voegen. Onder standaardomstandigheden geldt dat pH =
0.
Het Latimer-diagram voor chloor in een basische waterige oplossing (corresponderend
met pOH = 0 en bijgevolg pH = 14) wordt gegeven door:
Merk op dat de E°-waarde voor het redoxkoppel Cl2/Cl- in basisch milieu identiek is met de
waarde in zuur milieu, omdat de halfreactie geen protonentransfer inhoudt. De halfreacties
worden in evenwicht gebracht met OH- en H2O, omdat overwegend deze deeltjes in basisch
milieu aanwezig zijn. Zo is bijvoorbeeld de halfreactie voor ClO-/Cl2 koppel in basisch
milieu:
2 ClO-(aq) + 2 H2O(l) + 2e- → Cl2(g) + 4 OH-(aq)
E0 = +0,42 VDe
standaardpotentiaal van een koppel met deeltjes die niet naast elkaar staan in een Latimer-
25
diagram kan afgeleid worden uit de tussenliggende waarden door de potentialen om te zetten
in standaard Gibbs-energieën, door toepassing van de formule:ΔrG0 = -nFE0
(met F gelijk aan de Faraday-constante = 96485 Coulomb/mol en n is het aantal uitgewisselde
elektronen) en te steunen op het gegeven dat totale ΔrG0 waarde de som van de individuele
bijdragen is:ΔrG0 = ΔrG0’ + ΔrG0”
of na uitwerking en herschikking:
E =
0
n' E 0 ' + n" E 0"
n'+ n"
De reden waarom we de E0-waarden naar ΔrG0 –waarden omzetten, is omdat algebraïsche
regels op de ΔG-waarden toepasbaar zijn (wat niet het geval is voor de E0-waarden). Als
voorbeeld tonen we aan het het Latimer-diagram kan toegepast worden om de E0-waarde van
de reductie van HClO naar Cl- te berekenen.
Het oxidatiegetal van chloor verandert van +1 naar 0 in één stap (n’ = 1) en van 0 naar -1 in
een volgende stap (n" = 1). Door gebruik te maken van het Latimer-diagram kan men
schrijven:
HClO(aq) + H+(aq) + e- → 1/2Cl2(g) + H2O(l)
E0 = +1,67 V
1/2Cl2(g) + e- → Cl-(aq)
E0 = +1,36 V
Aan gezien n’ = 1 en n" = 1, is n = 2. Voor de E0-waarde van het koppel HClO/Cl- geldt:
E0 =
E 0 ' + E 0"
2
=
1,67V + 1,36V
= +1,51V
2
Aan de hand van de Latimer-diagrammen voor mangaan in zure oplossing (pH = 0,
corresponderend met een zuurconcentratie van 1 mol/L) en in basische oplossing (pH = 14,
corresponderend met een base-concentratie van 1 mol/L) kan men de zeer grote verschillen in
de redoxpotentialen voor deze twee voorwaarden goed aantonen:
26
De reden voor deze grote verschillen is te vinden in het groot aantal H+ of OH--ionen die
nodig zijn om de halfreacties in evenwicht te brengen. Elk effect dat een invloed op de
concentratie van deze ionen heeft, zal een zeer groot effect op de redoxpotentialen hebben.
Uit een Latimer-diagram kan men afleiden of een deeltje de neiging vertoont om te
disproportioneren. Beschouw de disproportioneringsreactie:
2M+(aq) → M(s) + M2+(aq)
Deze reactie verloopt spontaan als E0 > 0. Om dit criterium in termen van een Latimerdiagram te analyseren, kunnen we best de totale reactie als het verschil van twee halfreacties
uitdrukken:
M+(aq) + e- → M(s)
E0(R)
M2+(aq) + e- → M+(aq)
E0(L)
De aanduidingen L en R verwijzen naar de relatieve posities, links en rechts, van de koppels
in een Latimer-diagram (waarbij men moet onthouden dat de deeltjes met een hoge
oxidatietoestand aan de linkerzijde staan). De standaardredoxpotentiaal voor de totale reactie
is:
E0 = E0(R) – E0(L)
Deze uitdrukking is positief als E0(R) > E0(L). Hieruit kunnen we besluiten dat een deeltje de
thermodynamische neiging heeft om te disproportioneren in de naburige deeltjes als de E0waarde rechts van het deeltje in het Latimer-diagram groter is dan de E0-waarde links van het
deeltje. Een goed voorbeeld is H2O2, dat de neiging heeft om in zure omstandigheden in O2 en
H2O te disproportioneren:
0,70
1,76
O 2 ⎯+⎯
⎯→ H 2 O 2 ⎯+⎯
⎯→ H 2 O
27
Om dit te verifiëren, schrijven we de twee halfreacties uit:
H2O2(aq) + 2H+(aq) + 2e- → 2H2O(l)
E0 = +1,76 V = E0(R)
O2(g) + 2H+(aq) + 2e- → H2O2(aq)
E0 = +0,70 V = E0(L)
en maken we het verschil:
2H2O2(aq) → 2H2O(l) + O2(g)
E0 = +1,06 V
aangezien E0 > 0, zal de disproportionering spontaan verlopen. Merk op dat het Latimerdiagram niet kan vertellen of de disproportionering snel of traag zal verlopen, aangezien dat
met kinetische aspecten te maken heeft.
1.5.2 Frost-diagrammen
Een Frost-diagram is een plot van N E° voor het koppel X(N)/X(0) in functie van het
oxidatiegetal N van een element. Een Frost-diagram is nuttig om op een kwalitatieve manier
de relatieve stabiliteit van oxidatietoestanden weer te geven. Een andere benaming voor een
Frost-diagram is een volt-equivalent diagram. Een Frost-diagram kan beschouwd worden als
een 2D-versie van een Latimer-diagram. Een Frost-diagram kan eenvoudig uitgaande van het
overeenkomstige Latimer-diagram worden opgesteld. De waarden die op de Y-as moeten
worden uitgezet worden verkregen door het aantal elektronen die worden uitgewisseld tijdens
de verandering van oxidatietoestand te vermenigvuldigen met de standaardredoxpotentiaal
voor die verandering. Als voorbeeld wordt het Frost-diagram voor stikstof in Figuur 1.4
weergegeven.
28
Figuur 1.4. Frost-diagram voor stikstof. De volle lijn komt overeen met pH = 0
(standaardvoorwaarden), de gebroken lijn met pH = 14.
Om de constructie van een Frost-diagram te illustreren, beschouwen we het diagram voor
zuurstof. Het Latimer-diagram voor zuurstof is:
De oxidatiegetallen voor zuurstof zijn 0, -1 en -2 in de drie deeltjes. Voor de verandering van
oxidatiegetal van 0 naar -1 (van O2 naar H2O2) is E0 = +0,70 V en N = -1, dus NE0 = -0,70 V.
Omdat het oxidatiegetal van O in H2O -2 is, is N = -2 en omdat E0 voor vorming van H2O
+1,23 V is, is NE0 = -2,46 V. Met deze gegevens kunnen we het Frost-diagram voor zuurstof
tekenen (Figuur 1.5).
29
Figuur 1.5. Frost-diagram voor zuurstof.
Aangezien NE0 evenredig is met de standaard Gibbs-energie voor de omzetting van deeltje
X(N) in het element (NE0 = -ΔrG0/F), kan een Frost-diagram ook beschouwd worden als een
plot van de Gibbs energie in functie van het oxidatiegetal. De meest stabiele oxidatietoestand
van een element komt overeen met het deeltje dat onderaan in het Frost-diagram ligt (Figuur
1.6). De helling (richtingscoëfficiënt) van de verbindingslijn tussen twee punten in het Frostdiagram is gelijk aan de E0-waarde van het koppel verbonden door de rechte lijn. Hoe steiler
de helling is van de lijn tussen twee punten in een Frost-diagram, hoe hoger de potentiaal voor
het corresponderende koppel is (Figuur 1.7).
Figuur 1.6: Schematische voorstelling van de positie van de meest stabiele oxidatietoestand
in een Frost-diagram.
30
Figuur 1.7. Verband tussen de helling van de rechte in een Frost-diagram en
de waarde van de redoxpotentiaal.
De geoxideerde vorm in het koppel met de meest positieve helling zal reductie ondergaan. De
gereduceerde vorm in het koppel met de minst positieve helling zal oxidatie ondergaan
(Figuur 1.8).
Figuur 1.8. Relatieve stabiliteit van deeltjes in een Frost-diagram.
Een deeltje zal disproportioneren als de E0-waarde voor reductie van X(N) naar X(N-1) groter
is dan de E°-waarde voor de oxidatie van X(N) naar X(N+1). Een deeltje in een Frost-diagram
ondergaat spontane disproportionering als het deeltje gelegen is boven de verbindingslijn
tussen de deeltjes die als product van de disproportionering worden gevormd (Figuur 1.9).
Twee deeltjes hebben de neiging om spontaan te comproportioneren als het intermediair
deeltje lager ligt dan de verbindingslijn tussen de twee met elkaar reagerende deeltjes (Figuur
1.10).
31
Figuur 1.9. Disproportionering in een Frost-diagram.
Figuur 1.10. Comproportionering in een Frost-diagram.
Het gebruik van Frost-diagrammen kan aan de hand van dit voor mangaan worden
geïllustreerd (Figuur 1.11). Ten eerste kunnen we de oxiderende en reducerende deeltjes
identificeren. Deeltjes die hoog in het diagram liggen zijn oxiderende stoffen ten opzichte van
deeltjes die links ervan liggen, terwijl deze deeltjes die hoog in het diagram liggen als
reducerende stoffen optreden ten opzichte van andere oxiderende stoffen die rechts van hun
liggen. Deeltjes die in onderaan het Frost-diagram in een put liggen hebben een lage vrije
energie en ze vertonen weinig neiging om verder te reageren. Hieruit volgt dat MnO4- een
zeer sterk oxidans is. Terwijl er verschillende intermediaire oxidatietoestand kunnen
voorkomen is Mn2+ toch het meest stabiele deeltje dat in zuur milieu ontstaat door reductie
32
van MnO4-. Uit het Frost-diagram kan men ook afleiden dat Mn3+ zal disproportioneren tot
Mn2+ en MnO2.
Figuur 1.11. Frost-diagram voor mangaan in zuur milieu.
1.5.3 Pourbaix-diagrammen
Een Pourbaix-diagram (of E-pH-diagram) geeft de thermodynamische stabiele vorm van
een element weer als functie van de potentiaal en de pH-waarde. Zulk diagrammen geven een
visuele weergave van het oxiderende en reducerende vermogen van de belangrijkste stabiele
vormen van een element. Ze worden vaak gebruikt voor geochemische studies, evenals voor
milieu- en corrosiestudies. Een Pourbaix-diagram laat toe om de algemene relaties tussen
redox-activiteit en Brønsted-zuurheid te bestuderen. Het diagram toont aan onder welke
potentiaal- en pH-voorwaarden bepaalde deeltjes in water stabiel zijn. Pourbaix-diagrammen
zijn genoemd naar de elektrochemicus Marcel Pourbaix (1904-1998), die als professor aan de
Université Libre de Bruxelles verbonden was en die in 1938 deze diagrammen voor het eerst
heeft voorgesteld.
In Figuur 1.12 wordt een vereenvoudigd Pourbaix-diagram voor ijzer getoond. Om een
Pourbaix-diagram te lezen, moet men met een aantal gegevens rekening houden. Zo scheiden
de verticale lijnen deeltjes die met elkaar in een zuur-base-evenwicht staan, maar die niet
33
redox-actief zijn. De niet-verticale lijnen scheiden deeltjes die door redox-evenwichten met
elkaar verbonden zijn. De horizontale lijnen scheiden deeltjes in redox-evenwichten waarbij
geen protonen of hydroxide-ionen betrokken zijn. Schuine lijnen scheiden deeltjes in redoxevenwichten waarbij protonen of hydroxide-ionen betrokken zijn. Het stabiliteitsgebied van
water ten opzichte van oxidatie of reductie wordt door twee evenwijdige lijnen weergegeven.
Uit een Pourbaix-diagram kan men heel wat informatie afleiden. Elk punt in het diagram
geeft de thermodynamisch meest stabiele (en theoretisch de meest voorkomende) vorm van
een element bij de gegeven potentiaal- en pH-voorwaarden. Een Pourbaix-diagram geeft een
visuele weergave van de oxiderende en reducerende mogelijkheden van de belangrijkste
stabiele verbindingen van een element. De gebieden die door deze horizontale, verticale en
schuine volle lijnen worden afgebakend, zijn de overheersingsgebieden van de belangrijkste
deeltjes van een bepaald element. Sterk oxiderende deeltjes en oxiderende omstandigheden
worden enkel bovenaan een Pourbaix-diagram gevonden. De onderste stabiliteitsgrens van
sterk oxiderende deeltjes ligt ook hoog in het diagram. Sterk reducerende deeltjes en
reducerende omstandigheden worden onderaan in het diagram gevonden, en niet elders. Sterk
reducerende deeltjes hebben laaggelegen bovenste stabiliteitsgrenzen in het Pourbaixdiagram. Wanneer het overheersingsgebied voor een gegeven oxidatietoestand volledig
verdwijnt boven of beneden een gegeven pH-waarde en als het element in een intermediaire
oxidatietoestand is, zal het element disproportioneren. Een deeltje waarvan het
overheersingsgebied zich bij een gegeven pH uitstrekt van bovenaan tot onderaan het
Pourbaix-diagram, heeft geen oxiderende of reducerende eigenschappen bij die pH-waarde.
Pourbaix-diagrammen hebben echter ook een aantal beperkingen of tekortkomingen.
Aangezien deze diagrammen gebaseerd zijn op thermodynamische data, geven ze geen
informatie over de kinetica van corrosieprocessen. De diagrammen zijn enkel geldig voor een
specifieke temperatuur en voor welbepaalde drukvoorwaarden. De diagrammen zijn afgeleid
voor enkel een beperkt aantal concentraties van ionische deeltjes. De meeste Pourbaixdiagrammen beschouwen enkel zuivere verbindingen. Bijkomende berekeningen moeten
uitgevoerd worden als bijkomende deeltjes aanwezig zijn. Alhoewel een Pourbaix-diagram
toont dat bepaalde oxiden thermodynamisch stabiel zijn, hebben deze oxiden niet
noodzakelijk een beschermende (passiverende) werking.
34
Figuur 1.12. Vereenvoudigd Pourbaix-diagram voor enkele belangrijke in de natuur
voorkomende vormen van ijzer. De verticale streepjeslijn toont het normale pH-gebied in
natuurlijke wateren.
De constructie van een Pourbaix-diagram kan best geïllustreerd worden aan de hand
van het vereenvoudigd Pourbaix-diagram voor ijzer (Figuur 1.12). Beschouw eerst de
reductie-halfreactie voor het Fe3+/Fe2+-redoxkoppel:
Fe3+(aq) + e- → Fe2+(aq)
E0 = +0,77 V
Deze halfreactie bevat geen H+-ionen en bijgevolg is de potentiaal onafhankelijk van de pH,
zodat dit redox-evenwicht door een horizontale lijn in het Pourbaix-diagram wordt
weergeven. Als de omgeving een redoxkoppel met een potentiaal boven deze lijn (i.e. een
meer positief, meer oxiderend redoxkoppel) bevat, dan zal het geoxiderende deeltje, Fe3+, de
meest voorkomende vorm van ijzer in oplossing zijn. Bijgevolg is de lijn in de
linkerbovenhoek van het diagram een grens die de gebieden scheidt waar Fe3+ en Fe2+
domineren. Een andere belangrijke reactie is:
Fe3+(aq) + 3H2O(l) → Fe(OH)3(s) + 3H+(aq)
Deze reactie is geen redoxreactie, omdat geen enkel deeltje een verandering van
oxidatietoestand vertoont. De grens tussen het gebied waarin Fe3+ oplosbaar is en het gebied
waarin Fe(OH)3 neerslaat en Fe3+(aq) tot zeer lage concentraties beperkt is, is onafhankelijk
van om het even welk redoxkoppel. De grens is echter pH-afhankelijk, waarbij Fe3+(aq) wordt
35
begunstigd bij lage pH-waarden en Fe(OH)3 bij hoge pH-waarden. We nemen aan dat Fe3+ het
dominerende deeltje in oplossing is als de concentratie groter dan 10 μmol L-1 wordt (een
typische waarde voor ijzer in oppervlaktewateren). Bij lagere totale ijzerconcentraties komen
ook nog oligomere deeltjes met zuurstofbruggen voor, maar deze worden in het
vereenvoudigd diagram verwaarloosd. De evenwichtsconcentratie van Fe3+ varieert met de pH
en de verticale lijn in het Pourbaix-diagram geeft de pH weer waarbij Fe3+ het dominante
deeltje wordt volgens deze definitie. Een derde reactie die we moeten beschouwen, is de
volgende reductie:
Fe(OH)3(s) + 3H+(aq) + e- → Fe2+(aq) + 3H2O(l)
Deze reactie is pH-afhankelijk, aangezien H+ voorkomt in de halfreactie.
De pH-
afhankelijkheid kan expliciet door de Nernst-vergelijking worden weergegeven:
[Fe ]
[H ]
− (0,059 V ) log[Fe ] − (0,177 V ) pH
E = E 0 − (0,059 V ) log
=E
0
2+
+ 3
2+
De potentiaal daalt lineair als de pH stijgt, zoals weergegeven in Figuur 1.12. De lijn werd
getekend voor een Fe2+ concentratie van 10 μmol L-1. De gebieden met een potentiaal- en pHwaarde boven deze lijn komen overeen met omstandigheden waarin het geoxideerde deeltje
Fe(OH)3 stabiel is. De gebieden beneden deze lijn komen overeen met gebieden waarin het
gereduceerde deeltje Fe2+ stabiel is. Men kan uit het Pourbaix-diagram ook afleiden dat er in
zuur milieu sterkere oxiderende stoffen vereist zijn om Fe2+ te oxideren en neer te slaan dan in
basisch milieu. Het is een algemene regel voor gekoppelde redox/zuur-base-evenwichten dat
de aanwezigheid van H+ in het linkerlid van de halfreactie er voor zorgt dat de grens in het
Pourbaix-diagram een dalende rechte is, terwijl de aanwezigheid van H+ in het rechterlid van
de halfreactie er voor zorgt dat de grens een stijgende rechte is.
De verticale lijn bij pH = 9 verdeelt de gebieden waarbij ofwel de reactanten of de
producten stabiel zijn in de reactie:
Fe2+(aq) + 2H2O(l) → Fe(OH)2(s) + 2H+(aq)
Deze reactie is geen redoxreactie en de verticale lijn werd getrokken bij een pH-waarde die
correspondeert met een evenwichtsconcentratie van 10 μmol L-1 voor Fe2+. Een andere
verticale lijn scheidt de gebieden waarin Fe(OH)2 en Fe(OH)3 stabiel zijn:
36
Fe(OH)3(s) + H+(aq) + e- → Fe(OH)2(s) + H2O(l)
De potentiaal van deze redoxreactie is pH-afhankelijk, maar uit de Nernst-vergelijking volgt
dat de helling van de rechte minder steil is dan voor het Fe(OH)3/Fe2+ redoxkoppel (omdat er
een kleiner aantal H+-ionen bij betrokken is):
E = E 0 − (0,059 V ) log
1
H+
[ ]
= E − (0,059 V ) pH
0
Tenslotte kunnen we op het Pourbaix-diagram twee evenwijdige rechten tekenen om het
stabiliteitsgebied van water weer te geven. Zoals reeds hogerop vermeld, oxideert elk
redoxkoppel met een potentiaal die meer positief is dan de bovenste lijn van het
stabiliteitsgebied, water tot O2, en reduceert elk redoxkoppel met een potentiaal die meer
negatief is dan de onderste lijn van het stabiliteitsgebied, water tot H2. Alle redoxkoppels die
men moet beschouwen om de redoxreacties van ijzer in water te bespreken, liggen bijgevolg
in het stabiliteitsgebied van water.
Figuur 1.13. Overzicht van de E-pH-condities die heersen in verschillende typen van
oppervlaktewateren, met betrekking tot het stabiliteitsgebied van water.
Pourbaix-diagrammen laten toe om te voorspellen onder welke vormen de verschillende
elementen voorkomen in natuurlijke wateren. Wanneer oppervlaktewater in contact met de
atmosfeer komt, wordt het water met zuurstofgas verzadigd en vele redoxactieve deeltjes
37
kunnen door dit sterk oxidans worden geoxideerd (E° = +1,23 V). Meer volledig
gereduceerde deeltjes worden gevonden in de afwezigheid van zuurstof, zeker als er
organisch materiaal aanwezig is dat als reductans kan optreden. Het belangrijkste zuursysteem
dat de pH van het medium controleert, is het CO2/H2CO3/HCO3-/CO32- diprotische systeem,
waarbij atmosferisch CO2 voor het zuur zorgt en opgeloste carbonaatmineralen voor de base.
Biologische activiteit is ook van belang, omdat de ademhaling O2 opneemt en CO2 afgeeft.
Het zuuroxide CO2 doet de pH dalen en maakt bijgevolg de potentiaal positiever. Het
omgekeerde proces, de fotosynthese, verbruikt CO2 en zet O2 vrij. Dit verbruik van CO2 doet
de pH stijgen en maakt de absolute waarden van de potentialen kleiner. De potentiaal- en pHomstandigheden in typische natuurlijke oppervlaktewateren worden in Figuur 1.13
weergegeven. Door deze figuur te vergelijken met het Pourbaix-diagram van ijzer (Figuur
1.12), kunnen we afleiden dat Fe3+ in water kan bestaan als de omgeving oxiderend is. Als
bijgevolg O2 rijkelijk aanwezig is en als de pH laag is (pH < 4), zal ijzer als Fe3+ aanwezig
zijn. Omdat slechts weinig oppervlaktewaters zo zuur zijn, is het weinig waarschijnlijk dat
Fe3+ hierin kan gevonden worden. Ijzer in onoplosbaar Fe2O3 kan onder de vorm van Fe2+ in
oplossing gaan als er sterk reducerende omstandigheden aanwezig zijn. Men kan opmerken
dat bij stijging van de pH, Fe2+ enkel kan vormen in de aanwezigheid van sterk reducerende
redoxkoppels en dat bijgevolg de vorming van Fe2+ onwaarschijnlijk is in zuurstofrijke
wateren. Vergelijking van Figuur 1.12 met Figuur 1.13 toont aan dat ijzer zal gereduceerd
worden en opgelost worden onder de vorm van Fe2+ in water van veengebieden en in met
water verzadigde bodems die rijk aan organisch materiaal zijn. Het Pourbaix-diagram van
ijzer kan ook gebruikt worden om geofysische processen in water te begrijpen. Beschouw
bijvoorbeeld een meer waarbij een temperatuursgradiënt vermijdt dat er een verticale
menging van water plaatsvindt. Aan de oppervlakte is het water volledig door zuurstof
verzadigd en ijzer moet hier onder de vorm van onoplosbare Fe(OH)3-deeltjes aanwezig zijn.
Deze deeltjes hebben de neiging om te bezinken. Op grotere diepten is het zuurstofgehalte
van het water echter veel lager. Als er bovendien organisch materiaal of andere bronnen van
reducerende stoffen aanwezig zijn, zal Fe(OH)3 gereduceerd worden en het ijzer zal onder de
vorm van Fe2+ worden opgelost. De Fe2+-ionen zullen naar het wateroppervlak diffunderen,
waarbij ze met O2 in contact komen en terug tot het onoplosbare Fe(OH)3 worden geoxideerd.
Hierna kan de cyclus herbeginnen.
38
1.6 Corrosie van ijzer
De ongewenste (elektro)chemische aantasting van het oppervlak van metalen wordt in het
algemeen corrosie genoemd. De corrosie van ijzer en staal onder de vorming van een
oxidelaag heet roesten. Roestvorming treedt enkel op in de aanwezigheid van zuurstof en
water. Op een bepaalde plaats op het oppervlak van een ijzeren voorwerp, vindt de oxidatie
van het ijzer plaats:
Anode:
Fe(s) → Fe2+(aq) + 2 e-
Op een andere plaats op het oppervlak, vindt de reductie plaats. Hierbij is zuurstofgas en
water betrokken:
Kathode:
4 e- + O2(g) + 2 H2O → 4 OH-(aq)
Men kan beschouwen alsof er een miniatuur galvanische cel wordt gevormd, die wordt kort
gesloten. De elektronen die in het anodisch gebied worden gevormd, vloeien doorheen het
ijzer naar het kathodisch gebied. De kationen, de Fe2+-ionen, die aan de anode worden
gevormd, bewegen zich doorheen het water aan de oppervlakte van het ijzeren voorwerp naar
de kathode. De anionen, de OH--ionen, die aan de kathode worden geproduceerd, bewegen
zich naar de anode. De Fe2+ en de OH--ionen ontmoeten zich ergens tussen anode en kathode
en vormen Fe(OH)2. Ijzer(II)hydroxide is echter niet stabiel in de aanwezigheid van zuurstof
en vochtigheid. De verbinding wordt geoxideerd tot ijzer(III)hydroxide, dat in werkelijk
gehydrateerd ijzer(III) oxide, Fe2O3⋅xH2O, is. Dit gehydrateerd ijzer(III) oxide is bekend als
het roodbruine roest.
De plaatsen waar het roestende ijzeren voorwerp putjes vertoont (roestputjes of
roestpuntjes) zijn de anodische gebieden waar het ijzer onder de vorm van Fe2+ in oplossing
gaat. De kathodische gebieden zijn degene die het meest toegankelijk zijn voor zuurstof en
vocht, omdat O2 en H2O bij de kathodische reactie betrokken zijn. Roest vormt zich steeds op
plaatsen die verwijderd zijn van de plaatsen waar er putjes in het oppervlak worden gevormd
(tussen de kathodische en anodische gebieden).
Wanneer een geschilderd ijzeren voorwerp roestvorming vertoont, zijn de kathodische
gebieden deze waar de verf afgeschilferd is en het naakte ijzeroppervlak aan zuurstof en water
wordt blootgesteld. De anodische gebieden waar het ijzer putjesvorming vertoont, zijn
plaatsen onder het geschilderde oppervlakte. De putjesvorming zorgt ervoor dat er meer verf
afschilfert, waardoor de roestvorming nog wordt versneld. De roest vormt zich op plaatsen
tussen deze twee gebieden, maar vaker dichter bij het kathodische dan bij het anodische
39
gebied, aangezien voor de omvorming van Fe(OH)2 tot roest O2 en H2O vereist zijn. De
putvorming in het ijzer kan leiden tot verlies van de materiaalsterkte. De grootste schade
veroorzaakt door corrosie is niet de zichtbare roestvorming, maar de schade die optreedt
beneden het geschilderde oppervlak.
Bij aanwezigheid van zwaveldioxide (afkomstig van industriegassen die in de atmosfeer
worden uitgestoten) wordt ijzer(II)sulfaat gevormd, dat zich als zgn. “sulfaatnesten” in de
laag ijzerroest afzet. In een eerste stap wordt geadsorbeerd SO2 tot H2SO4 geoxideerd, waarbij
roest als katalysator voor de reactie optreedt:
SO2 + ½ O2 + H2O → H2SO4
Dit H2SO4 kan ijzer aantasten en vormt FeSO4 en water:
Fe + H2SO4 + ½ O2 → FeSO4 + H2O
Ook reeds neergeslagen ijzer(II)hydroxide kan dan in ijzer(II)sulfaat worden omgezet:
Fe(OH)2 + SO42- → FeSO4 + 2OH-
Zulke ijzersulfaten zijn sterk roest bevorderend. Zij veroorzaken een sterke diepte-inwerking
van het roestproces, waaroor putjes in staal ontstaan (de zgn. putvormige corrosie). Het in
roest aanwezige ijzersulfaat kan als volgt worden omgezet:
4 FeSO4 + 6 H2O + O2 → 4 FeO(OH) + 4H2SO4
Daardoor
worden
sulfaationen
voor
verdere
roestvorming
vrijgemaakt.
De
ijzer(II)sulfaatnesten vervullen derhalve de rol van opslagplaats voor sulfaationen.
Zout water versnelt de roestvorming omdat de Na+ en Cl- ionen die in het water aanwezig
zijn het water meer geleidend maken en ervoor zorgen dat de elektrische stromen in de
miniatuur galvanische cel aan de oppervlak van het ijzer gemakkelijker kunnen lopen. Het is
bekend dat ijzerroest relatief grote hoeveelheden natriumchloride kan opnemen, onder
vorming van het onstabiele oplosbare ijzer(II)chloride. Het gevormde ijzer(II) chloride kan
dan in ijzeroxidehydraat en waterstofchloride worden omgezet.
Fe2+ + 2Cl- → FeCl2
40
4FeCl2 + 6H2O + O2 → 4FeO(OH) + 8 HCl
Dit waterstofchloride zal opnieuw ijzer in oplossing doen gaan onder de vorming van
ijzer(II)chloride, zodat de roestsnelheid aanzienlijk wordt vergroot. De versnelde corrosie
door zout water is heel duidelijk bij auto’s als er in de winter zout op de wegen tegen
ijzelvorming wordt gestrooid. Ook zeewater veroorzaakt snellere corrosie, waardoor
corrosiebescherming in de scheepsbouw een belangrijk probleem is. Een verhoogde zuurheid
leidt tot versnelde corrosie; vandaar dat er water verzadigd met CO2 of zure regen voor sterke
corrosie zorgen. Onzuiverheden in het ijzer versnellen ook de roestvorming. Zeer zuiver ijzer
roest niet snel. In droge lucht en in lucht- en koolstofdioxidevrij water wordt compact ijzer
niet aangetast. Deze bestendigheid is terug te brengen door de aanwezigheid van een
aaneengesloten dunne oxidelaag. Dankzij deze oxidelaag wordt ijzer ook niet aangetast door
geconcentreerd zwavelzuur of geconcentreerd salpeterzuur, zodat ijzeren vaten kunnen
gebruikt worden voor het transport van deze geconcentreerde zuren. Deze dunne
beschermende oxidelaag staat in schril contrast met een roestlaag. Een roestlaag vormt geen
aaneengesloten laag, maar schilfert af, waardoor er verse ijzeroppervlakken worden
blootgesteld en de roestvorming verder in het ijzeren voorwerp kan binnendringen. Door het
ruwe, poreuze, roestige oppervlak wordt het water meer opgenomen en hierdoor wordt het
roestproces in de hand gewerkt. Het volume van roest is groter dan het volume van het ijzer
waaruit de roest is ontstaan. Als een ijzerdeeltje in roest overgaat, wordt zijn volume ongeveer
tweemaal zo groot. Is ter bescherming tegen roesen het ijzer of staal met een verflaag bedekt,
dan kan door het ontstaan van roest de verflaag scheuren en afschilferen. Ijzer de opgesloten
zit in metselwerk , beton of natuursteen, kan wanneer het gaat roesten aan de bouwconstructie
grote beschadigingen veroorzaken.
Ijzeren of stalen voorwerpen kunnen tegen roestvorming worden beschermd door een
beschermlaag (vet, olie, verf, plastic, email of andere metalen) op het oppervlak aan te
brengen. Deze beschermlaag verhindert het contact van ijzer met O2 en H2O. Metaalcoatings
kunnen door elektrolyse worden aangebracht (bijv. chroom of nikkel) of door het voorwerp in
een bad van gesmolten metaal te dompelen (bijv. zink of tin). Gegalvaniseerd ijzer is ijzer dat
met een zinklaagje bedekt is. Het ijzer van gegalvaniseerd ijzer is tegen roestvorming
beschermd, zelfs al raakt de zinklaag beschadigd. In gegalvaniseerd ijzer treedt zink eerder
dan ijzer als anode op. Zink wordt geoxideerd, aangezien zink reactiever dan ijzer is.
Bovendien vormt het zink bij corrosie een onoplosbare film van Zn(OH)2 op het oppervlakte,
waardoor verdere corrosie wordt vertraagd. Voor ijzer dat met een laagje tin wordt beschermd
(bijv. conserveblikjes) is het omgekeerde waar. Als het tinlaagje beschadigd geraakt, wordt de
corrosie van het onderliggende ijzer versneld, aangezien ijzer reactiever dan tin is. Vroeger
werd ijzer vaak tegen corrosie beschermd door middel van een verf op basis van
41
lood(IV)oxide (loodmenie, Pb3O4) die zorgde voor oppervlakkige oxidatielaag en passivering
van het ijzer. Omwille van milieuredenen mag loodmenieverf niet meer worden gebruikt.
Staallegeringen met een hoog gehalte aan chroom zijn roestvast. Roestvrij staal bevat
minimum
10,5%
chroom
en
maximaal
1,2%
koolstof,
naast
eventuele
andere
legeringsmetalen. Het chroom vormt een onzichtbaar laagje chroomoxide (Cr2O3) wanneer
het in aanraking komt met zuurstof, de oxidehuid. Dit laagje beschermt het onderliggende
metaal tegen verdere roestvorming (oxidatie). Bovendien herstelt het zichzelf wanneer het
beschadigd wordt. Het is echter een foutieve veronderstelling dat roestvrij staal niet kan
roesten. In omstandigheden die hardnekkig genoeg zijn, of bij beschadiging van de
beschermende oxide-huid, kan de roestvorming plaatselijk extra snel plaatsvinden. Dit wordt
o.a. veroorzaakt door chloriden of andere metalen die zich nestelen in het oppervlak. Dit is
ook de reden waarom bij het bewerken van roestvrij staal bij voorkeur geen stalen
gereedschappen gebruikt zouden mogen worden. Praktischer is het echter om na het
verwerken, het roestvrij te behandelen waarbij alle mogelijke verontreinigingen worden
verwijderd. Dit is het zogenaamde beitsen, waarbij langs chemische weg alle
ongerechtigheden worden opgelost en verwijderd. Bepaalde soorten roestvrij staal bevatten
naast chroom ook aanzienlijke hoeveelheden nikkel. Zo is inox 18/2 een ijzerlegering met
18% chroom en 8% nikkel. Roestvrij staal van het type 316 bevat wat minder chroom en iets
meer nikkel en minimaal 2% molybdeen. Het wordt heel vaak toegepast in de chemische
industrie omwille van zijn zeer hoge corrosiebestandigheid. De term edelstaal wordt gebruikt
om hoogwaardig roestvrij staal aan te duiden dat onder andere gebruikt wordt voor
keukengerei en bestek.
Een andere vorm van bescherming tegen corrosie is kathodische bescherming. Onder
kathodische bescherming verstaat men het tegengaan of voorkomen van corrosie van metalen
in waterig milieu of in elektroliet door middel van metalen die in de gegeven omstandigheden
onedeler zijn dan het te beschermen metaal. Metalen als aluminium, zink en magnesium
komen in aanmerking voor de bescherming van ijzer. Deze metalen offeren zich als het ware
voor het te beschermen metaal op. Daarom worden de anoden die uit deze metalen worden
vervaardigd dan ook opofferingsanoden genoemd. Om een ondergrondse ijzeren tank,
pijpleiding of kabel tegen roestvorming te beschermen, worden stukken van reactieve metalen
zoals magnesium of zink naast het ijzeren voorwerp begraven en door middel van metalen
draden met het ijzeren voorwerp verbonden. Hierdoor wordt het ijzer niet geoxideerd: het
wordt de kathode en het reactievere metaal wordt de anode. De reactieve metaalanoden
worden opgeofferd om het ijzer te beschermen. De opofferingsanoden oxideren snel weg en
moeten regelmatig worden vervangen. Ook voor de bescherming van scheepsrompen worden
offerelektroden gebruikt. Voor kathodische bescherming moeten de opofferingsanode en het
te beschermen ijzeren voorwerp voortdurend in contact zijn met water of met door water
42
verzadigde grond. Een tweede vorm van kathodische bescherming kan worden verkregen
door het zenden van een elektrische stroom door het te beschermen metaal door middel van
een uitwendige stroombron. Met behulp van een gelijkrichter wordt een negatieve spanning
aan het metaal aangebracht, die een elektronische stroom doet ontstaan die verhindert dat het
te beschermen metaal in oplossing gaat.
1.7 Metallurgische processen
1.7.1 Ellingham-diagrammen
Ondanks het feit dat redoxreacties niet altijd hun evenwichtstoestand bereiken, kan men
toch van thermodynamica gebruik maken om uit te maken of een reactie al dan niet spontaan
verloopt. Het criterium voor het spontane verloop van een reactie onder de heersende
omstandigheden (bij een constante druk en temperatuur), is dat de Gibbs vrije energie van de
reactie negatief is. Het is gewoonlijk voldoende om de standaard Gibbs vrije energie ΔG0, te
nemen, omdat deze in verband staat met de evenwichtsconstante K via de volgende
vergelijking:
ΔG0 = - RT ln K
(1.10)
Bijgevolg komt een negatieve waarde voor ΔG0 overeen met een K > 1, en daarom met een
spontane reactie. Reactiesnelheden zijn ook relevant, maar bij hoge temperaturen zijn reacties
gewoonlijk toch snel, zodat men kan veronderstellen dat bij hoge temperaturen elke
thermodynamisch gunstig proces zal opgaan.
Om een negatieve ΔG0 te verkrijgen voor de reductie van een metaaloxide met koolstof of
met koolstofmonoxide, moet één van de volgende reacties:
(a) C(s) + ½O2(g) → CO(g)
ΔG0(C,CO)
(b) ½C(s) + ½O2(g) → ½CO2(g)
ΔG0(C,CO2)
(c) CO(g) + ½O2(g) → CO2(g)
ΔG0(CO,CO2)
een meer negatieve ΔG0-waarde hebben dan een reactie van de vorm:
(d) xM(s of l) + ½O2(g) → MxO(s)
ΔG0(M,MxO)
43
onder dezelfde reactieomstandigheden. Als dat zo is dan zal één van de onderstaande reacties
een negatieve Gibbs vrije energie hebben en bijgevolg spontaan verlopen:
(a-d) MxO(s) + C(s) → xM(s of l) + CO(g)
ΔG0(C,CO) - ΔG0(M,MxO)
(b-d) MxO(s) + ½C(s) → xM(s of l) + ½CO2(g)
ΔG0(C,CO2) - ΔG0(M,MxO)
(c-d) MxO(s) + CO(g) → xM(s of l) + CO2(g)
ΔG0(CO,CO2) - ΔG0(M,MxO)
De relevante informatie over dit typen van reacties wordt gewoonlijk grafisch in een
Ellingham-diagram samengevat. Een Ellingham-diagram is een plot van de standaard Gibbs
vrije energie van de vorming van oxiden als functie van de temperatuur (Figuur 1.14). De
vorm van een Ellingham kan begrepen worden door op te merken dat:
ΔG0 = ΔH0 -TΔS0
(1.11)
en door gebruik te maken van het gegeven dat de enthalpie ΔH0 en de entropie ΔS0 van een
reactie grotendeels temperatuursonafhankelijk zijn. De helling van een rechte in een
Ellingham-diagram is bijgevolg gelijk aan -ΔS0 voor de relevante reactie. Omdat de
standaardentropie van een gas veel groter is dan deze van een vaste stof, is de
standaardentropie van reactie (d), waarbij er een netto verbruik van gassen is, negatief, en
bijgevolg heeft de plot van deze reactie in het Ellingham-diagram een positieve helling. De
knikken in de rechten, waarbij de helling van de lijn van de metaaloxidatie verandert, zijn
gelegen bij temperaturen waarbij het metaal een faseverandering ondergaat, en dan vooral bij
het smeltproces, en waar de reactie-entropie verandering ondergaat. De reactie-entropie van
reactie (a), waarbij er een netto vorming van een gas optreedt (omdat 1 mol CO, ½ mol O2
vervangt) is positief, waardoor de overeenkomstige lijn in het Ellingham-diagram een
negatieve helling heeft. De standaardreactieentropie van reactie (b) is nagenoeg gelijk aan nul,
omdat er geen netto-verandering in de hoeveelheid gas optreedt, waardoor de lijn in het
Ellingham-diagram een horizontale rechte is. Reactie (c) heeft een negatieve reactie-entropie,
omdat 3/2 mol gasmoleculen wordt vervangen door 1 mol CO2. Hierdoor heeft de
overeenkomstige reactie in het Ellingham-diagram een positieve helling.
44
Figuur 1.14. Variatie van de standaard Gibbs vrije energie voor de vorming van een
metaaloxide en koolstofmonoxide als functie van de temperatuur. De vorming van
koolstofmonoxide uit koolstof is in staat om het metaaloxide tot metaal te reduceren bij
temperaturen die hoger zijn dan het snijpunt van de twee lijnen. Bij het snijpunt verandert de
waarde van de evenwichtsconstante K van kleiner dan 1 tot groter dan 1.
Figuur 1.15. Een fragment van een Ellingham-diagram toont de standaard Gibbs energie
voor de vorming van een metaaloxide en voor de drie oxidatiereacties waarbij koolstof
betrokken is. De hellingen van de rechten worden grotendeels bepaald door het feit of er
netto-vorming of netto-verbruik van gassen tijdens de reactie gebeurt. Een faseverandering
resulteert in een knik in de grafiek (omdat de entropie van de verbinding verandert).
45
Bij temperaturen waarbij de C,CO-lijn boven de metaaloxidelijn in Figuur 1.16 ligt, is
ΔG (M,MxO) negatiever dan ΔG0(C,CO). Bij deze temperaturen, is ΔG0(C,CO) 0
ΔG0(M,MxO) positief, zodat de reactie (a-d) niet spontaan is. Voor temperaturen waarbij de
C,CO-lijn beneden de metaaloxidelijn ligt, is de reductie van het metaaloxide door koolstof
spontaan. Gelijkaardige besluiten kunnen getrokken worden voor temperaturen waarbij de
andere twee koolstofoxidatielijnen boven of beneden de metaaloxidelijn in Figuur 1.15
liggen. Samenvattend kan men zeggen:
•
Voor temperaturen waarbij de C,CO-lijn onder de metaaloxidelijn ligt, kan koolstof
worden gebruikt om het metaaloxide te reduceren en wordt koolstof zelf tot
koolstofmonoxide geoxideerd.
•
Voor temperaturen waarbij de C,CO2-lijn onder de metaaloxide lijn ligt, kan koolstof
worden gebruikt om het metaaloxide te reduceren en wordt het zelf tot
koolstofdioxide geoxideerd.Voor temperaturen waarbij de CO,CO2-lijn onder de
metaaloxide lijn ligt, kan koolstofmonoxide worden gebruikt om het metaaloxide te
reduceren en wordt het zelf tot koolstofdioxide geoxideerd.Als ΔG0(M,MxO) bij een
bepaalde temperatuur positief wordt, is vorming van metaal uit oxide mogelijk zonder
toevoeging van reductans.
In Figuur 1.16 wordt een Ellingham-diagram voor een selectie aan metalen getoond. Uit
dit diagram kan bijvoorbeeld worden afgeleid dat de laagste temperatuur waarbij zink uit
zinkoxide door koolstof kan worden gereduceerd ongeveer 950 °C bedraagt. Boven deze
temperatuur ligt de (C,CO)-lijn onder de (Zn,ZnO)-lijn en verloopt de reductie van ZnO met
C spontaan. Bij deze temperatuur bevindt zinkmetaal zich in de gasvormige toestand,
aangezien het kookpunt van zink 907 °C is. Het kookpunt van zink is waar te nemen als een
knik in de (Zn,ZnO)-lijn. In principe zijn alle metalen die in het diagram worden getoond,
zelfs magnesium en aluminium, door verhitting met een reducerend reagens uit het
metaaloxide te verkrijgen. Er bestaan echter serieuze praktische beperkingen. Zo kan het
moeilijk en duur zijn om de vereiste zeer hoge temperaturen te bereiken. Pogingen om titaan
te bereiden via de pyrometallurgische methode leiden tot de vorming van titaancarbide in
plaats van titaanmetaal. De methode kan worden toegepast voor de bereiding van ijzer, kobalt,
nikkel, zink, magnesium en voor een hele reeks ferrolegeringen. Meer informatie wordt in de
volgende sectie gegeven.
46
Figuur 1.16. Ellingham-diagram voor de reductie van metaaloxiden. De weergegeven
standaard Gibbs vrije energieën zijn weergegeven voor de vorming van metalen uit
metaaloxiden en voor de drie koolstofoxidatiereacties.
Gelijkaardige principes zijn van toepassing voor andere reducerende reagentia dan
koolstof. Zo kan het Ellingham-diagram bijvoorbeeld gebruikt worden om af te leiden of een
metaal M’ kan gebruikt worden om het oxide van een andere metaal M te reduceren. Men
moet controleren bij welke temperatuur de (M’,MxO’)-lijn beneden de (M,MxO)-lijn ligt, want
M’ neemt nu de plaats van C in. Bijvoorbeeld, de (Mg,MgO)-lijn ligt onder de (Si,SiO2)-lijn
voor temperaturen lager dan 2200 °C, zodat magnesium beneden deze temperatuur kan
gebruikt worden om SiO2 tot Si te reduceren. Deze methode wordt trouwens in de praktijk
gebruikt om siliciummetaal van een lagere zuiverheid te bereiden.
1.7.2 Pyrometallurgie
De winning van metalen uitgaande van ertsen noemt men extractieve metallurgie. De
vroegst bekende metalen zijn goud, zilver en koper, omdat deze elementen in de Natuur ook
in elementvorm (gedegen toestand) voorkomen. De meeste metalen komen echter voor in
47
ertsen onder de vorm van verbindingen met zuurstof of zwavel, en er zijn chemische
methoden nodig om deze metalen uit hun ertsen vrij te maken. Uit Tabel 1.1 kan men afleiden
dat de vrije vormingsenergie van de meeste metaaloxiden negatief is. Dit betekent dat de
omgekeerde reacties die het vrije metaal en zuurstof leveren een positieve verandering van de
vrije energie inhoudt en bijgevolg niet spontaan verlopen. Men kan de omgekeerde reactie ter
vorming van een vrij metaal enkel laten verlopen door deze reactie te koppelen met een
tweede, spontaan verlopende reactie. Hoe negatiever de waarden voor de vrije
vormingsenergie zijn, des te moeilijker zal de productie van het vrije metaal zijn. Goud en
zilver staan onderaan in Tabel 1.1 en komen zoals vermeld in de Natuur in gedegen toestand
voor. Gelijkaardige informatie kan men uit Ellingham-diagrammen afleiden (zie hogerop).
Vele metalen worden geproduceerd door middel van reductieprocessen op hoge
temperatuur, waarbij de metalen in gesmolten toestand worden verkregen. Dit zijn de
zogenaamde smeltprocessen (of pyrometallurgische processen). Bij de meeste processen
wordt een vloeimiddel of flux (meestal kalksteen, CaCO3) gebruikt om het ganggesteente te
verwijderen dat na concentratie van het erts overblijft. De flux vormt een slak met
siliciumdioxide en silicaatonzuiverheden. Voor kalksteen en siliciumdioxide (kwarts) is de
vereenvoudigde vergelijking:
CaCO3(s) → CaO(s) + CO2(g)
CaO(s) + SiO2(s) → CaSiO3(l)
De slak is vloeibaar bij de temperatuur waarbij het smeltproces wordt uitgevoerd, drijft
gewoonlijk bovenop het gesmolten metaal en kan gemakkelijk van het metaal worden
afgescheiden.
De reducerende stof die voor een bepaald smeltproces wordt gebruikt is het goedkoopste
materiaal dat in staat is om een product met gewenste zuiverheid te verkrijgen. Voor ertsen
van metalen met een lage reactiviteit, zoals voor de sulfidische ertsen van kwik, koper of lood
zijn geen chemische reductiemiddelen vereist. Kwik kan verkregen worden door cinnaber,
HgS, in lucht te verhitten:
HgS(s) + O2(g) → Hg(g) + SO2(g)
De kwikdamp wordt in een opvangvat gecondenseerd en vereist meestal geen verdere
zuivering. Het metallurgisch proces waarbij men een sulfidisch erts in de aanwezigheid van
lucht verhit, noemt men roosten.
48
Sulfidische koperertsen worden eerste opgesmolten tot een materiaal dat men matte
noemt en dat grotendeels uit koper(I)sulfide, Cu2S, bestaat. De matte wordt dan tot koper
gereduceerd (met gelijktijdige oxidatie van het sulfide tot SO2) door lucht door het gesmolten
materiaal te blazen:
Cu2S(l) + O2(g) → 2Cu(l) + SO2(g)
Het koper dat op deze manier wordt gevormd heeft een zuiverheid van ongeveer 99% en
wordt door middel van elektrolyse verder gezuiverd.
Loodsulfide-erts (galeniet) wordt aan een roostproces onderworpen waarbij een gedeelte
van loodsulfide tot loodoxide en loodsulfaat wordt omgezet:
2PbS(s) + 3O2(g) → 2PbO(s) + 2SO2(g)
PbS(s) + 2O2(g) → PbSO4(s)
Het resulterende mengsel van PbS, PbO en PbSO4 wordt samen met een vloeimiddel
gesmolten in de afwezigheid van lucht. De reacties waarbij zwavel wordt geoxideerd en lood
wordt gereduceerd zijn:
PbS(s) + 2PbO(s) → 3Pb(l) + SO2(g)
PbS(s) + PbSO4(s) → 2Pb(l) + 2SO2(g)
Een deel lood wordt ook geproduceerd door PbO samen met cokes te verhitten. Hierbij wordt
het lood(II)oxide verkregen door het loodsulfide-erts in een overmaat lucht te verhitten zodat
er omzeggens een volledige omzetting tot PbO plaats vindt.
Ijzer, nikkel, kobalt, molybdeen, zink, cadmium, tin, antimoon en andere metalen worden
geproduceerd door reductie met koolstof van de oxiden die verkregen worden als ertsen of als
producten van het roostproces van sulfidische ertsen. De reacties die optreden bij
koolstofreductie op hoge temperatuur zijn complex. In de meeste gevallen wordt de reductie
echter door koolstofmonoxide en niet door koolstof verwezenlijkt. Aangezien zowel het
metaaloxide als koolstof (onder de vorm van cokes) vaste stoffen zijn die niet gemakkelijk
smeltbaar zijn, is het contact tussen deze reagentia slecht en de directe reactie traag:
MO(s) + C(s) → M(l) + CO(g)
49
(M staat voor een metaal). Tussen een vaste stof en een gas is er echter een beter contact
mogelijk en deze reagentia reageren sneller:
2C(s) + O2(g) → 2CO(g)
MO(s) + CO(g) → M(l) + CO2(g)
Koolstofdioxide dat wordt gevormd wordt tot koolstofmonoxide omgezet door reactie met
cokes:
C(s) + CO2(g) → 2CO(g) + 221,20 kJ
Pyrometallurgische processen zijn niet in staat om metalen te verkrijgen die bovenaan in
Tabel 1.1 staan. Deze elementen hebben zeer grote negatieve vrije vormingsenergieën voor
hun oxiden (en hun sulfiden). De beste methode om deze metalen te winnen is via
elektrometallurgie (elektrolytische winning). Merk op dat elektrometallurgische processen
ook worden gebruikt voor de zuivering van metalen die via pyrometallurgische processen
worden geproduceerd.
Tabel 1.1. Rangschikking van metaaloxiden volgens de gemakkelijkheid van reductie
Metaaloxide
Metaal
n
ΔG0f /n (kJ/mol)
MgO
Mg
2
-285
Al2O3
Al
6
-264
TiO2
Ti
4
-222
Na2O
Na
2
-188
Cr2O3
Cr
6
-176
ZnO
Zn
2
-159
SnO2
Sn
4
-130
Fe2O3
Fe
6
-124
NiO
Ni
2
-106
PbO
Pb
2
-94
CuO
Cu
2
-65
HgO
Hg
2
-29
Ag2O
Ag
2
-6
Au2O3
Au
6
>0
50
ΔG0f is de standaard vrije energie voor de vorming van de metaaloxiden, uitgedrukt in kJ/mol.
Om een vergelijking mogelijk te maken worden de waarden gedeeld door n, zijnde de totale
daling in oxidatietoestand vereist om de metaalatomen in het oxide te reduceren tot
oxidatietoestand 0. Bijgevolg houdt de reductie van Cr2O3 de verandering in van de
oxidatietoestand van twee chroomatomen van +3 naar 0, zodat n = 2 × 3 = 6.
1.7.3 Productie van ijzer
Ijzer is het nuttigste metaal en wordt bijgevolg in reusachtige hoeveelheden bereid. Het
komt in de Natuur slechts zeer zelden in gedegen vorm voor, maar het komt onder de vorm
van oxiden, sulfiden, silicaten en carbonaten zeer verspreid en in zeer grote hoeveelheden
voor. De voor de ijzerindustrie belangrijkste ijzerertsen zijn magnetiet (Fe3O4), hematiet
(Fe2O3), limoniet (Fe2O3⋅xH2O) en sideriet (FeCO3). Pyriet (FeS2) kan pas tot ijzer worden
verwerkt, als het eerst geroost wordt. Het gevormde zwaveldioxide doet dan dienst als
grondstof voor de productie van zwavelzuur.
De metallurgie van ijzer in in beginsel zeer eenvoudig. Ze berust op de reductie van
ijzeroxiden tot ijzer op hoge temperatuur. Dit proces wordt uitgevoerd in een hoogoven, die
ontworpen is om continu in werking te blijven (Figuur 1.17). Een typische hoogoven heeft
een hoogte van 25 tot 30 meter, een diameter van ongeveer 10 meter en een volume van 500
tot 800 m3. De hoogoven is uit vuurvaste steen opgebouwd en bestaat in essentie uit twee
kegelvormige schachten, die met hun basis samenvallen. Jaarlijks kan één hoogoven ca. 1
miljoen ton ijzer produceren. Een hoogoven wordt slechts om de ongeveer 10 jaren stilgelegd
als de binnenbekleding vervangen moet worden. De hoogoven wordt constant verhit en de
grondstoffen worden continue bovenaan de oven toegevoegd, terwijl het gesmolten ijzer en de
gevormde slakken onderaan worden afgetapt.
De ijzerertsen worden vaak eerst geroost om de vluchtige stoffen (H2O, CO2, S, As, ...) te
verwijderen en om hen losser te maken. Vervolgens worden de ijzerertsen verbrokkeld en
naargelang hun ijzergehalte met de nodige slakkenvormende stoffen, de “toeslag” gemengd.
Als het ganggesteente rijk aan siliciumdioxide of aluminiumoxide is, dan verkiest men als
bijmengsel kalksteen, CaCO3, of dolomiet, CaMg(CO3)2. Als echter het ijzererts veel calciumof magnesiumoxide bevat, wordt kwarts (siliciumdioxide) voor de slakkenvorming
toegevoegd. Het ijzererts, cokes en de toeslag worden bovenaan in de oven geladen. Verhitte
lucht, die soms met zuurstof is aangerijkt, wordt onderaan in de oven geblazen. De
binnenstromende lucht reageert met koolstof ter vorming van koolstofdioxide:
C(s) + O2(g) → CO2(g)
51
Deze reactie is zeer exotherm en er komen hierbij aanzienlijke hoeveelheden warmte vrij. Op
deze plaats is de temperatuur van de oven het hoogste en de temperatuur kan tot 2000 °C
oplopen. Door de overmaat aan zuurstof wordt er op deze plaats in de hoogoven wel CO2,
maar geen CO gevormd. CO2 reageert vervolgens met hete koolstof ter vorming van
koolstofmonoxide:
CO2(g) + C(s) ' 2CO(g)
Dit is een evenwichtsreactie, en wordt de Boudouard-reactie genoemd. Het evenwicht schuift
naar
rechts
bij
toenemende
temperatuur,
zodat
bij
hogere
temperaturen
meer
koolstofmonoxide wordt gevormd.
Het ijzererts wordt stapsgewijze gereduceerd, afhankelijk van de temperatuur. Nabij
de bovenkant van de oven, waar de temperatuur het laagste is, wordt er eerst Fe3O4
(magnetiet) gevormd:
3Fe2O3(s) + CO(g) → 2Fe3O4(s) + CO2(g) + 43,7 kJ
Deze reactie treedt op rond 500 °C. Het gevormde Fe3O4 zakt in de oven naar beneden en
wordt in de lager gelegen, hetere zone tot FeO (wüstiet) gereduceerd:
Fe3O4(s) + CO(g) → 3FeO(s) + CO2(g) – 36,8 kJ
Deze reactie treedt op rond 850 °C. In de zone tussen 1000 °C en 1200 °C wordt FeO door
CO tot metallisch ijzer gereduceerd:
FeO(s) + CO(g) → Fe(l) + CO2(g) + 17,2 kJ
De ontbinding van kalksteen tot calciumoxide in het midden van de hoogoven verloopt
volgens de reactie:
CaCO3 → CaO + CO2
Het gevormde calciumoxide reageert met verschillende zure onzuiverheden in het ijzer
(vooral met siliciumdioxide) ter vorming van slakken die grotendeels uit calciumsilicaat,
CaSiO3, bestaan.
52
Figuur 1.17. Schematische voorstelling van een hoogoven.
Het gesmolten ijzer verzamelt zich onderaan de hoogoven. De gesmolten slakken, die
vooral bestaan uit calciumsilicaat dat gevormd wordt door de reactie van het vloeimiddel met
het ganggesteente, verzamelt zich ook onderaan de oven. De slakken drijven op het gesmolten
ijzer, waardoor het ijzer wordt beschermd tegen de lucht die boven de laag gesmolten slakken
in de hoogoven wordt geblazen. De gesmolten slakken en het gesmolten ijzer worden
geregeld afgetapt. De gestolde slakken kunnen naargelang hun samenstelling gebruikt worden
bij de wegenbouw of voor de vervaardiging van cement. De hoeveelheid slakken is
vergelijkbaar met de geproduceerde hoeveelheid ijzer. De hete uitlaatgassen worden gebruikt
om de binnnenkomende lucht in de hoogoven te verhitten.
Het onzuivere ijzer dat wordt geproduceerd in een hoogoven, noemt men ruwijzer. Het
bevat koolstof (2,5-4%), silicium (0,5-3%), mangaan (0,5-6%), fosfor (0-2%) en sporen van
zwavel (0,01%-0,05%). Men onderscheidt twee vormen van ruwijzer, naargelang of het
silicium- dan wel het mangaangehalte overheerst. Als het ruwijzer veel silicium bevat en zeer
langzaam afkoelt (bijv. in zandvormen), dan scheidt een groot gedeelte van de opgeloste
koolstof zich als grafiet uit en men verkrijgt dan zogenaamd grijs ruwijzer, met een grijs
53
breukvlak (smeltpunt ∼1200 °C). Dit siliciumhoudende grijze ruwijzer wordt omwille van zijn
lage viscositeit in gesmolten vorm vooral gebruikt voor het vervaardigen van gegoten ijzeren
werkstukken. Hiertoe wordt het voor gebruik nog een tweede maal opgesmolten. Men noemt
grijs ruwijzer daarom ook gietijzer. Als men het ruwijzer echter snel laat afkoelen (bijv. in
een ijzeren ketel), dan blijft koolstof onder de vorm van ijzercarbide, Fe3C (“cementiet”) in
het ijzer opgelost. Men verkrijgt dan een wit ruwijzer, met een wit breukvlak (smeltpunt ∼
1100 °C). Wit ruwijzer vereist een vrij groot mangaangehalte (> 4% Mn), omdat mangaan de
uitscheiding van koolstof als grafiet tegenwerkt. Het witte ruwijzer wordt verder tot staal of
smeedijzer verwerkt. De reden waarom koolstof bij traag afkoelen niet in ijzer opgelost blijft,
is omdat cementiet enkel bij hoge temperaturen stabiel is, en bij traag afkoelen in ijzer en
grafiet uiteenvalt.
Het ruwijzer is wegens zijn verhoudingsgewijze hoge koolstofgehalte (tot 4%) broos en
het wordt bij verhitting niet geleidelijk, maar plots zacht. Het kan bijgevolg niet gesmeed of
gelast worden. Om ruwijzer in een smeedbare vorm om te vormen, moet men het
koolstofgehalte tot beneden 1,7% verlagen. De smeedbare vormen van ijzer met een
koolstofgehalte tussen 0,5 en 1,7%, noemt men staal. Het is moeilijk smeltbaar, maar plet- en
smeedbaar. Men kan verschillende stukken bij hoge temperatuur, waarbij het staal in een
deegachtige toestand overgaat, door hameren of walsen tot één stuk verenigen. Het smeltpunt
van staal is des te hoger naarmate de bijmengingen afnemen. Als men het staal op een
bepaalde, met zijn koolstofgehalte samenhangende temperatuur (750 °C - 900 °C) verhit en
dan snel in water of olie afgekoelt, dan wordt het zeer hard (harden van staal). Smeedbaar
ijzer met een koolstofgehalte lager dan 0,5% wordt smeedijzer genoemd. Het is zeer moeilijk
smeltbaar en wordt als het witgloeiend is (voor zijn overgang tot de vloeibare toestand) week,
deegachtig en is dan gemakkelijk aan elkaar te wellen. Dit smeedijzer is niet meer te harden.
Zuiver ijzer met minder dan 0,1% koolstof wordt ook wel vloeiijzer genoemd. Het
onderscheid tussen staal en smeedijzer is niet zo heel scherp. Vaak worden alle
ijzerlegeringen met een koolstofgehalte lager dan 1,7% staal genoemd. Kleine bijmengingen
van andere elementen oefenen soms een zeer nadelige invloed op de eigenschappen van ijzer
uit. Zwavel maakt reeds in kleine hoeveelheden het ijzer broos in de hitte en dus ongeschikt
om te smeden. Vandaar dat zwavelhoudende ijzerertsen als dusdanig voor de ijzerfabricage
ongeschikt zijn. Fosfor maakt ijzer bij gewone temperaturen broos.
Uit het ruwijzer, zoals het uit de hoogoven komt, worden de andere ijzersoorten bereid.
Het moet daartoe ontdaan worden van silicium, mangaan, zwavel, fosfor en voor een groot
gedeelte ook van zijn koolstofgehalte. Het technische proces waarbij dit het snelste wordt
bereikt, maar dat tegenwoordig door modernere processen is verdrongen, is het Bessemerproces. Het gesmolten ruwijzer zoals het uit de hoogoven komt, wordt in een peervormig, om
54
een middenas draaibaar toestel, de converter, gegoten. In de bodem van de converter zijn er
openingen waardoor lucht wordt ingeblazen. Daarbij wordt, zonder gebruik van brandstof,
door de oxidatie van silicium, mangaan en een klein gedeelte ijzer, de temperatuur hoog
genoeg opgevoerd om ook de verbranding van koolstof mogelijk te maken. Bij dit proces
komen zeer hoge temperaturen voor, waarbij 1650 °C een gemiddelde waarde is. Nadat dit is
gebeurd, voegt men spiegelijzer of ferromangaan toe. Dit zijn legeringen van ijzer en
mangaan met een gehalte van respectievelijk 20 en 30-80% mangaan, die in hoogovens uit
een mengsel van ijzer en mangaanertsen op dezelfde manier als ruwijzer verkregen worden.
Het mangaan reduceert de in het eerste gedeelte van het proces gevormde ijzeroxiden weer tot
ijzer en neemt bovendien de nog aanwezige zwavel op. Daar de legering ook betrekkelijk veel
koolstof bevat, wordt bereikt dat het eindproduct weer een zeker gehalte aan koolstof heeft.
Het Thomas-proces is een variant van het Bessemer-proces, dat voor fosforhoudend ijzer
wordt toegepast. Als men fosforhoudend ijzererts gebruikt, dan moet de converter een
basische bekleding bestaande uit dolomiet, CaMg(CO3)2, of magnesiet, MgCO3, en moet men
aan het gesmolten ruwijzer nog kalksteen toevoegen. Het gevormde P2O5 wordt dan aan CaO
en MgO gebonden en de gevormde fosfaten worden door het gesmolten ijzer niet meer
gereduceerd. Als bijproduct verkrijgt men een basische fosfaathoudende slak. Tot fijn poeder
vermalen is dit het zogenaamde thomasfosfaatmeel, dat veel als fosforhoudende meststof
wordt gebruikt. Een moderner proces dan het Bessemer-proces is het Linz-Donawitzer-proces.
Dit proces wordt ook het LD-proces of het basisch- zuurstof-proces (naar de basische
bekleding van de converter met calcium- en magnesiumoxiden) genoemd. Bij het LinzDonawitzer-proces wordt een mengsel van gesmolten ruwijzer en ijzerschroot in de converter
geladen. Vervolgens wordt zuurstofgas onder druk (7-10 atm.) door middel van een water
gekoelde zuurstoflans op het gesmolten ijzeroppervlak geblazen. De hevige verbranding
(oxidatie) van de ijzerbijmengingen zorgt er voor dat de gesmolten ijzermassa gemengd
wordt. Voor een betere menging kan ook argongas door openingen onderaan de converter
worden geblazen. Het opblazen van zuurstofgas duurt tussen 10 minuten en 20 minuten totdat
het gewenste gehalte aan koolstof en onzuiverheden wordt bereikt. De geoxideerde
ijzerbijmengingen ontwijken als gassen of worden door op dat moment toegevoegde
kalksteen tot vloeibare slakken gebonden. Vervolgens wordt de converter gekanteld en het
gesmolten staal wordt in een gigantische kuip uitgegoten. Hier kunnen dan eventuele
legeringsmetalen zoals mangaan, nikkel chroom of vanadium aan het staal worden
toegevoegd. Als het staal uit de converters is afgetapt, worden de vloeibare slakken uit de
converter gegoten.
55
1.7.4 Elektrometallurgie: Productie van aluminium
Aluminium is het derde meest voorkomende element in de aardkorst (na zuurstof en
silicium), en vormt ongeveer 8,2% van de totale massa van de aardkorst. Het komt
gewoonlijk samen met silicium voor in aluminosilicaten zoals veldspaten en glimmers, maar
ook in kleien die het verweringsproduct van aluminosilicaatgesteenten zijn. Het belangrijkste
erts voor de winning van aluminium is echter bauxiet. Bauxiet is een gehydrateerd
aluminiumoxide dat bestaat uit 50% tot 60% Al2O3, 1% tot 20% Fe2O3, 1% tot 10% SiO2,
kleinere concentraties aan oxiden van titaan, zirkonium, vanadium en andere transitiemetalen,
en 20% tot 30% water. Mineralogisch gezien is bauxiet een gesteente dat overwegend bestaat
uit hydrargilliet, Al(OH)3, diaspoor, α-AlO(OH) en böhmiet, γ-AlO(OH), met bijmengingen
van kleimineralen. Grote bauxietafzettingen komen voor in Frankrijk, Hongarije, Verenigde
Staten, Guayana, China en Australië. Bauxiet wordt gezuiverd via het Bayer-proces dat
voordeel haalt uit het feit dat het amfotere aluminiumoxide oplosbaar is in sterke basen, maar
ijzer(III)oxide niet. Ruw bauxiet wordt in een autoklaaf onder hoge druk (5-7 atm) en hoge
temperatuur (170-178 °C) in een geconcentreerde natriumhydroxide-oplossing opgelost:
Al2O3(s) + 2OH-(aq) + 3H2O(l) → 2[Al(OH)4]-(aq)
en door filtratie afgescheiden van gehydrateerd ijzeroxide en andere onoplosbare
onzuiverheden. Zuiver gehydrateerd aluminiumoxide slaat neer als een verzadigde oplossing
wordt afgekoeld en door toevoeging van kristalkiemen tot kristallisatie wordt gebracht:
2[Al(OH)4]-(aq) → Al2O3⋅3H2O(s) + 2OH-(aq)
Merk op dat Al2O3⋅3H2O ook als 2Al(OH)3 kan weergegeven worden. Het hydratatiewater
wordt door calcinatie op hoge temperatuur (1200 °C) verwijderd:
2Al(OH)3(s) → Al2O3(s) + 3H2O(g)
Het in bauxiet aanwezige kiezelzuur gaat bij de behandeling met natriumhydroxide-oplossing
grotendeels over in onoplosbaar natriumaluminiumsilicaat, Na2[Al2SiO6]⋅2H2O,
SiO2 + 2 NaOH + Al2O3 → Na2[Al2SiO6] + H2O
Dit slaat samen met ijzer(III)hydroxide neer.
56
In vergelijking met koper, ijzer, goud, zilver en lood die reeds in de Oudheid gekend
waren, is aluminium een relatief nieuw metaal. Sir Humphrey Davy verkreeg aluminium als
een ijzerlegering en toonde de metallische aard van aluminium in 1809 aan. Aluminium werd
eerst in onzuivere vorm verkregen in 1825 door H.C. Oersted door de reductie van
aluminiumchloride met een kaliumamalgaam:
AlCl3(s) + 3K(Hg)x(l) → 3KCl(s) + Al(Hg)3x(l)
Vervolgens werd kwik door destillatie verwijderd. Friedrich Wöhler verkreeg in 1827 zuiver
aluminium door de directe reductie van aluminiumchloride met kalium. Aluminium bleef een
laboratoriumcuriosum tot 1886 wanneer onafhankelijk van elkaar Charles Martin Hall in de
Verenigde Staten en Paul Héroult in Frankrijk een efficiënt proces voor de
aluminiumproductie ontwikkelden. Jaarlijks wordt er wereldwijd meer dan 15 miljoen ton
aluminium via het Hall-Héroult-proces geproduceerd.
Het Hall-Héroult-proces houdt de kathodische afzetting van aluminium in, in
elektrolysecellen die gesmolten cryoliet (Na3AlF6) met opgelost Al2O3 bevatten. Het voor de
elektrolyse gebruikte aluminiumoxide moet zeer zuiver zijn. Elke elektrolysecel bestaat uit
een rechthoekige stalen tank die ongeveer 6 meter lang, 2 meter breed en 1 meter hoog is, en
die als de kathode dienst doet. De anoden bestaan uit grafietstaven die in het gesmolten
cryolietbad steken. Enorme elektrische stromen (50000 tot 100000 A) worden door de
elektrolysecel geleid, maar het potentiaalverschil is echter laag (3-5 V).
Gesmolten cryoliet is volledig gedissocieerd in Na+ en [AlF6]3- ionen. Het is een excellent
oplosmiddel voor aluminiumoxide. Een elektrolysebad bestaande uit gesmolten cryoliet en
opgelost aluminiumoxide bevat een evenwichtsverdeling aan ionen zoals Al3+, AlF2+, ....,
[AlF6]3- en O2-. Cryoliet smelt op 1000 °C, maar het smeltpunt wordt lager door het opgeloste
aluminiumoxide, zodat de werkingstemperatuur van de elektrolysecel ongeveer 950 °C
bedraagt. Dit is een lage temperatuur in vergelijking met het smeltpunt van zuiver Al2O3
(2050 °C) and dit is de reden waarom het Hall-Héroult-proces een commercieel succes is
geworden. Gesmolten aluminium heeft een iets grotere massadichtheid dan het gesmolten
zoutbad bij 950 °C en verzamelt zich bijgevolg op de bodem van de elektrolysecel, waar het
regelmatig kan worden afgetapt. Onder in het elektrolysebad is het gesmolten aluminium
beschermd tegen terugoxidatie door zuurstof. Zuurstof is een primair anodeproduct, maar het
reageert met de grafietelektrode ter vorming van koolstofdioxide, dat onder de vorm van
gasbellen uit het elektrolysebad ontwijkt. De totale celreactie is:
2Al2O3 + 3C → 4Al + 3CO2
57
De bovenkant en de zijkanten van het elektrolysebad zijn bedekt met een laag vast cryoliet,
dat voor een thermische isolatie zorgt. De elektrische weerstand van het elektrolysebad is
voldoende hoog om het cryoliet in gesmolten toestand te houden. Meer dan 100
elektrolysecellen kunnen in serie worden geplaatst. De productie van 1 ton aluminium vereist
ongeveer 65-70 GJ (18-20 MWh) aan energie en ongeveer 500 kg koolstof. Het Hall-Héroultproces is niet vervuilend, maar het verbruikt zeer veel elektriciteit. Slechts ongeveer 36% van
de electriciteit wordt voor Faraday-processen gebruikt, terwijl de rest onder de vorm van
warmte verloren gaat.
Aluminium en zijn legeringen hebben zeer veel toepassingen. Vele toepassingen
maken gebruik van de lage massadichtheid van aluminium (2,35 g/cm3), dat een groot
voordeel is ten opzichte van ijzer en staal wanneer gewichtsbesparing wenselijk is (bijv. voor
auto’s). Aluminium is een goede geleider van elektriciteit en is goedkoper dan koper. Het
wordt daarom gebruikt voor elektriciteitsdraden in hoogspanningsleidingen. Aluminium is
ook interessant voor toepassingen bij zeer lage temperaturen, omdat aluminium bij lagere
temperaturen sterker wordt terwijl ijzer en staal onder deze omstandigheden vaak broos
worden. Huishoudfolie en drankblikjes kunnen dankzij de lage toxiciteit van aluminium uit
dit metaal worden vervaardigd.
58
Hoofdstuk 2:
Metaalclusters
De termen “cluster” en “metaalcluster” worden vaak als synoniemen gebruikt, alhoewel
de respectievelijke definities duidelijk verschillen. Een “cluster” is een opeenstapeling van
drie of meer atomen van eenzelfde element of van gelijkaardige elementen waarbij er directe
chemische bindingen tussen deze elementen bestaan. Een “metaalcluster” wordt daarentegen
gedefinieerd als een verbinding met metaal-metaalbindingen. Ook een bimetallische
verbinding met een metaal-metaalbinding wordt een metaalcluster genoemd. Het is af te raden
om de term “cluster” te gebruiken voor meerkernige coördinatieverbindingen waarbij geen
direct metaal-metaalbinding voorkomt.
Verbindingen
met
metaal-metaalbindingen
zijn
reeds
lang
bekend.
Zo
werd
kwik(I)chloride, Hg2Cl2, bekend onder de benaming calomel, reeds vanaf de middeleeuwen
als laxeer- en ontsmettingsmiddel gebruikt. De dimere aard van het Hg22+ werd pas rond het
einde van de 19de eeuw bevestigd. Vanaf ongeveer 1960 werden metaalclusters intensief
onderzocht. Men kan twee grote klassen van metaalclusters onderscheiden, namelijk deze van
het “carbonyl”-type en het “halide”-type. Vele metalen zijn in staat om metaalclusters te
vormen. Nochtans hebben de metalen met de hoogste atomisatie-energie (en dus met het
hoogste smeltpunt en kookpunt) zoals Zr, Nb, Mo, Tc, Ru, Rh, Hf, Ta, W, Re, Os, Ir en Pt de
grootste neiging om metaalclusters te vormen.
2.1 Isolobale moleculaire fragmenten
Een belangrijk concept om de chemische binding in metaalclusters te begrijpen is het
concept van isolobale moleculaire fragmenten, dat werd geïntroduceerd door de Amerikaanse
chemicus en nobelprijswinnaar Roald Hofmann. De definitie luidt:
Moleculaire fragmenten zijn isolobaal als het getal, de symmetrie-eigenschappen, de
benaderde energie en de vorm van de grensorbitalen en het aantal elektronen in deze
orbitalen gelijkaardig is ⎯ niet identiek, maar gelijkaardig.
Om deze definitie te illustreren is het nuttig om de fragmenten van methaan te vergelijken met
de fragmenten van een octaëdrisch gecoördineerd transitiemetaalcomplex ML6. Voor de
eenvoud beschouwen we enkel de σ-bindingen tussen het metaal en de liganden in dit
complex. In Figuur 2.1 worden de tetraëdrische en octaëdrische fragmenten die besproken
worden, getoond.
59
Figuur 2.1. Orbitalen van octaëdrische en tetraëdrische fragmenten.
De basisverbindingen hebben een gevulde valentieschil, zijnde een octetconfiguratie voor
CH4 en 18 elektronen voor ML6. Een voorbeeld van zulke een metaalcomplex is Cr(CO)6.
Men mag aannemen dat methaan sp3-hybride-orbitalen gebruikt voor zijn chemische
bindingen, waarbij 8 elektronen de bindende moleculaire orbitalen bezetten die gevormd
worden tussen de sp3-hybriden en de 1s orbitalen van waterstof. Het metaal in ML6 maakt
gebruikt van d 2 sp 3 -hybriden voor de chemische bindingen met de liganden, waarbij 12
elektronen de bindende moleculaire orbitalen bezetten en 6 elektronen zich bevinden in
moleculaire orbitalen die grotendeels niet-bindend zijn, namelijk de dxy-, dxz- en dyz-orbitalen.
We kunnen nu moleculaire fragmenten beschrijven die minder liganden bevatten dan de
boven beschreven fragmenten. Hierbij neemt men aan dat de geometrie van de overblijvende
liganden bewaard blijft. In het CH3-fragment met 7 elektronen zijn drie van de sp3-orbitalen
van koolstof betrokken in de σ-binding met de waterstofatomen. De vierde hybride-orbitaal is
enkelvoudig bezet en ligt bij een hogere energie dan de σ-bindende moleculaire orbitalen van
CH3. Deze situatie is gelijkaardig met de binding in het Mn(CO)5-fragment dat 17 elektronen
heeft. Vijf van de d 2 sp 3 -orbitalen van mangaan zijn betrokken in de σ-interacties tussen de
liganden en het metaal. De zesde hybride-orbitaal bevat één elektron en heeft een hogere
energie dan de vijf bindende moleculaire orbitalen met σ-symmetrie. Figuur 2.1 toont dat elk
60
van deze fragmenten een ongepaard elektron heeft in een hybride-orbitaal op een lege plaats
in de coördinatiepolyeder van de basisverbinding. Hoffmann voerde een speciaal symbool in
. Men kan dus schrijven: CH3 ←⎯→
ML5 om uit te
om isolobale groepen aan te duiden: ←⎯→
O
O
drukken dat de CH3- en ML5-fragmenten isolobaal zijn. Op een gelijkaardige wijze zijn CH2
met 6 elektronen en ML4 met 16 elektronen isolobaal. Zowel CH2 als ML4 hebben twee
elektronen te kort voor een gevuld octet- of een 18-elektronenconfiguratie, zodat ze
elektronisch equivalent zijn. Beide hebben twee hybride-orbitalen die door een ongepaard
elektron bezet zijn op sites die voor de rest leeg zijn. De afwezigheid van een derde ligand
geeft het paar van isolobale fragmenten CH en ML3, waarbij telkens drie hoekpunten
ontbreken in de coördinatiepolyeder van de basisstructuur en drie elektronen te kort zijn voor
een gevulde schil. In Figuur 2.2 worden de isolabaliteitsrelaties nogmaals getoond.
Figuur 2.2. Isolobaliteitsrelaties tussen de fragmenten van CH4 en ML6.
Deze isolobale fragmenten kunnen tot moleculen worden gecombineerd. Bijvoorbeeld,
twee CH3-fragmenten vormen ethaan en twee Mn(CO)5 fragmenten vormen het dimeer
(OC)5Mn⎯Mn(CO)5. Bovendien kunnen deze twee fragmenten worden gecombineerd tot
H3C⎯Mn(CO)5 (Figuur 2.3).
61
Figuur 2.3. Combinaties van de isolobale fragmenten CH3 en Mn(CO)5.
De parallellen tussen organische fragmenten en organometaalfragmenten gaan niet altijd
volledig op. Bijvoorbeeld, terwijl twee CH2-fragmenten het stabiele molecule etheen,
H2C=CH2, vormen, is het dimeer van het isolobale fragment Fe(CO)4 onstabiel. Zowel CH2
als Fe(CO)4 vormen echter drieringen, namelijk cyclopropaan en Fe3(CO)12. Alhoewel
cyclopropaan een trimeer is van drie CH2-fragmenten, heeft Fe3(CO)12 twee brugvormende
carbonylgroepen en is bijgevolg geen perfect trimeer van Fe(CO)4. Het iso-elektronische
Os3(CO)12 is echter wel een trimeercombinatie van drie Os(CO)4-fragmenten, die isolobaal
zijn met Fe(CO)4 en CH2, en de verbinding kan correct worden geschreven als [Os(CO)4]3.
Het concept van isolobale fragmenten kan uitgebreid worden naar geladen deeltjes, naar
een waaier aan liganden naast CO en naar organometaalfragmenten die een andere structuur
hebben dan een octaëder. Bijvoorbeeld, aangezien het Mn(CO)5-fragment met 17 elektronen
isolobaal is met het CH3-fragment met 7 elektronen, zijn ook de fragmenten Re(CO)5,
[Fe(CO)5]+ en [Cr(CO)5]- die elk 17 elektronen hebben, isolobaal met CH3. Het verlies of
opname van elektronen van twee isolobale fragmenten levert nieuwe isolobale fragmenten.
Bijvoorbeeld de fragmenten Cr(CO)5, Mo(CO)5 en W(CO)6 die elk 16 elektronen hebben, zijn
isolobaal met CH3+, dat 6 elektronen heeft. Fe(CO)5, Ru(CO)5, Os(CO)5 die elk 18 elektronen
hebben, zijn isolobaal met CH3- dat een fragment met 8 elektronen is. Alle hierboven
vermelde fragmenten hebben één ligand te kort in vergelijking met de coördinatiepolyeder
van de basisstructuur. Fe(CO)5 en CH4 zijn echter niet isolobaal. Beide hebben een gevulde
elektronenschil (met resp. 18 en 8 elektronen), maar bij CH4 zijn alle hoekpunten van de
tetraëder bezet, terwijl Fe(CO)5 een lege positie heeft in de octaëder. Andere tweeelektrondonoren kunnen op dezelfde manier als het CO-ligand beschouwd worden:
62
Mn(PR3)5 ←⎯→
[MnCl5]5- ←⎯→
CH3
Mn(CO)5 ←⎯→
O
O
O
Men beschouwt dat η5-C5H5 en η6-C6H6 drie plaatsen bezetten in de coördinatiepolyeder en
dat ze 6-elektrondonoren zijn:
(η5-C5H5)Fe(CO)2 ←⎯→
[Fe(CO)5]+ ←⎯→
Mn(CO)5
O
O
(fragmenten met 17 elektronen)
(η6-C6H6)Mn(CO)2
(η5-C5H5)Mn(CO)2 ←⎯→
[Mn(CO)5]+ ←⎯→
Cr(CO)5
O
O
(fragmenten met 16 elektronen)
(η6-C6H6)Cr(CO)2
Octaëdrische fragmenten met formule MLn (waarbij M een d x -configuratie heeft) zijn
isolobaal met vierkant-vlakke fragmenten met formule MLn-2 (waarbij M een d x +2configuratie heeft en L een 2-elektrondonor is):
Octaëdrische fragmenten
Vierkant-vlakke fragmenten
MLn
Cr(CO)5
MLn-2
←⎯→
O
[PtCl3]-
d6
d8
Fe(CO)4
Pt(PR3)2
d
8
d10
Een vergelijk tussen de octaëdrische en de vlak-vierkante fragmenten wordt in Figuur 2.4
getoond. Een vierkant-vlak ML3-fragment met een d 8 -configuratie (zoals [PtCl3]-) heeft als
LUMO een lege lob van een niet-bindende hybride-orbitaal. Deze is vergelijkbaar met de
LUMO van een octaëdrisch ML5-fragment met een d 6 -configuratie (bijv. Cr(CO)5):
63
Een d 8 -fragment zoals [PtCl3]- is isolobaal met Cr(CO)5 en andere ML5-fragmenten op
voorwaarde dat de lege lob in beide gevallen een geschikte energie heeft. Een ML2-fragment
met d10-configuratie zoals Pt(PR3)2 heeft twee elektronen meer dan het voorbeeld dat in
Figuur 2.4 wordt getoond. Men veronderstelt dat deze twee elektronen twee niet-bindende
hybride-orbitalen bezetten. Deze situatie is erg vergelijkbaar met deze van het Fe(CO)4fragment (Figuur 2.1). Elk complex heeft twee enkelvoudig bezette lobben:
Figuur 2.4. Vergelijking tussen vierkant-vlakke fragmenten en octaëdrische fragmenten.
Deze analogieën zijn niet beperkt tot octaëdrische en vierkant-vlakke complexen.
Gelijkaardige argumenten kunnen worden gebruikt om isolobale fragmenten van
verschillende polyeders af te leiden. Bijvoorbeeld, Co(CO)4 is een fragment van een trigonale
bipiramide met 17 elektronen en is isolobaal met Mn(CO)5, een fragment van een octaëder
met 17-elektronen:
64
Het concept van isolobale fragmenten kan uitgebreid worden naar om het even welk
moleculair fragment met grensorbitalen van een geschikte grootte, vorm, symmetrie en
energie. Bijvoorbeeld, Au(PPh3) is een fragment met 13 elektronen en heeft een ongepaard
elektron in een hybride-orbitaal die van fosfine-liganden wegwijst. Dit elektron bezet een
orbitaal met een gelijke symmetrie, maar met een iets hogere energie dan de hybride-orbitaal
met een ongepaard elektron in het fragment Mn(CO)5. Desalniettemin kan Au(PPh3) zich
combineren
met
de
isolobale
fragmenten
Mn(CO)5
en
CH3
ter
vorming
van
(OC)5Mn⎯Au(PPh3) en H3C⎯Au(PPh3).
Zelfs een waterstofatoom met één enkel elektron in een 1s-orbitaal kan in sommige gevallen
beschouwd worden als een fragment dat isolobaal is met deeltjes zoals CH3, Mn(CO)5 en
Au(PPh3). Bijvoorbeeld, H en Mn(CO)5 vormen het hydride HMn(CO)5. De belangrijkste
mogelijke toepassing van het concept van isolobale fragmenten is het herkennen van
mogelijke nieuwe organometaalverbindingen die zouden kunnen gesynthetiseerd worden.
2.2 Metaal-metaalbindingen
Het concept van isolobale verbindingen werd in de vorige sectie gebruikt om de vorming
van metaal-metaalbindingen te beschrijven, zoals in het voorbeeld carbonylcomplex
(OC)5Mn⎯Mn(CO)5. Metaal-metaalbindingen verschillen van andere bindingen enkel in het
gebruik van de d-orbitalen op beide metalen. Naast de gewone σ- en π-bindingen zijn bij
transitiemetalen ook δ-bindingen mogelijk. Hierdoor zijn transitiemetalen in staat om
viervoudige bindingen met elkaar te vormen (zie 2.3). Omdat er daarnaast ook brugvorming
tussen twee metalen met liganden en de mogelijkheid om clusterverbindingen te vormen
optreden, zorgen er voor dat er een brede waaier bestaat aan verbindingen met metaalmetaalbindingen. De structuur van carbonylcomplexen met metaal-metaalbindingen werd
besproken in de cursus Metalen en Katalyse.
65
2.3 Meervoudige metaal-metaalbindingen
Transitiemetalen kunnen met elkaar enkelvoudige, dubbele, drievoudige, viervoudige
bindingen vormen, en ook bindingen met een niet-gehele bindingsorde. Men kan zich
afvragen hoe viervoudige bindingen mogelijk zijn. Bij de hoofdgroepelementen kunnen
moleculaire orbitalen met elkaar overlappen op een σ- of π-wijze. Wanneer twee
hoofdgroepmetaalatomen met elkaar interageren is de hoogste mogelijke bindingsorde drie,
zijnde een combinatie van een σ-binding en twee π-bindingen. Wanneer twee
transitiemetaalatomen met elkaar interageren, vindt de belangrijkste interactie plaats via de dvalentie-orbitalen. Deze d-orbitalen reageren ter vorming van moleculaire orbitalen die niet
alleen een σ- of π-symmetrie hebben, maar ook met een δ-symmetrie (Figuur 2.5).
Figuur 2.5. Bindingsinteracties tussen d-orbitalen van metalen.
Als de z-as als de internucleaire as wordt gekozen, is de sterkste interactie (met de grootste
orbitaal-overlap) de σ-interactie tussen de dz2-orbitalen. De volgende in sterkte van de
interactie is de overlap van de dxz- en dyz-orbitalen die π-orbitalen vormen als resultaat van
interacties in twee gebieden in de ruimte. De laatste en zwakste interactie is deze tussen de
dxy- en dx2-y2-orbitalen, die interageren in vier gebieden in de ruimte, ter vorming van
66
moleculaire orbitalen met δ-symmetrie. De relatieve energie van de resulterende moleculaire
orbitalen wordt schematisch in Figuur 2.6 weergeven. Een M2-fragment zou in de
afwezigheid van liganden vijf bindende moleculaire orbitalen vormen door de d-d interacties,
waarbij de energie van deze moleculaire orbitalen toeneemt volgens σ < π < δ < δ* <π* < σ*.
Figuur 2.6. Relatieve energieën van moleculaire orbitalen die gevormd worden door de
interactie tussen d-orbitalen.
De klassieke voorbeelden complexen met meervoudige metaal-metaalbindingen zijn de
[Re2X8]2- ionen. Ze kunnen bereid worden door reductie van het perrenaat-ion met H2 of
H3PO2 in de aanwezigheid van X--ionen (X = Cl, Br, I, NCS):
2ReO4- + H3PO2 + HX → [Re2X8]2-
Het meest interessante aspect van deze verbindingen is hun structuur, die twee ongewone
kenmerken vertoont. Dit wordt in Figuur 2.7 voor het [Re2Cl8]2- ion getoond. De eerste is de
67
ongewoon korte Re⎯Re afstand van 224 pm, in vergelijking met een gemiddelde Re⎯Re
van 275 pm in reniummetaal. Het tweede onverwachte kenmerk is de geëclipseerde
conformatie van de chlooratomen (D4h-symmetrie). Aangezien de korte Re⎯Re afstand
vereist dat de chlooratomen op afstanden van elkaar liggen (∼330 pm) die kleiner zijn dan de
som van hun van der Waalsstralen (340-360 pm), verwacht men dat de gestaggerde
conformatie (waarbij de chlooratomen een vierkant antiprisma in plaats van een kubus
vormen) zou verkozen worden. De Amerikaanse chemicus F. Albert Cotton (1930-2007)
verklaarde beide fenomen door aan te nemen dat er een viervoudige binding tussen de
reniumatomen wordt gevormd.
Figuur 2.7. Structuur van het octachlorodirenaat(III) ion, [Re2Cl8]2-.
Voor de eenvoud beschouwen we dat de z-as is gelegen volgens de lijn die de twee
reniumatomen verbinden en dat de Re-Cl-bindingen in de xz- en yz-vlakken gelegen zijn. Elk
reniumatoom is gebonden aan vier chlooratomen in een bijna vierkant-vlakke coördinatie (elk
Re-atoom ligt ongeveer 50 pm boven het vlak gevormd door de vier Cl-atomen). De
ligandorbitalen interageren het sterkste met de metaalorbitalen die naar hen gericht zijn, in dit
geval de δ- en δ*-orbitalen die grotendeels van de dx2-y2-orbitalen afkomstig zijn. Het gevolg
van deze interactie is dat er nieuwe moleculaire orbitalen worden gevormd, zoals getoond
wordt op de rechterzijde van Figuur 2.7. De relatieve energieën van deze orbitalen is sterk
afhankelijk van de sterkte van de metaal-ligandinteracties, die variëren van complex tot
complex. In [Re2Cl8]2- is elk reniumatoom formeel in de oxidatietoestand +3 en heeft vier delektronen. Als de acht d-elektronen voor dit ion in de vier moleculaire orbitalen met de
laagste energie worden geplaatst zoals die getoond zijn in figuur 2.6 (met uitsluiting van de
moleculaire orbitalen met lage energie die grotendeels van dx2-y2-interacties afkomstig zijn en
door ligandelektronen worden bezet), dan is de totale bindingsorde 4, corresponderend met (in
toenemende energie) een σ-binding, twee π-bindingen en een δ-binding. De δ-binding is de
68
zwakste binding, maar ze is sterk genoeg om het ion in een geëclipseerde conformatie te
dwingen. De zwakte van de δ-binding wordt geïllustreerd door het kleine energieverschil
tussen de δ- en δ*-orbitalen. Het energieverschil komt typisch overeen met de energie van
zichtbaar licht. Bijgevolg hebben de meeste complexen met een viervoudige binding een
intense kleur. Bijvoorbeeld, [Re2Cl8]2- is donkerblauw en [Mo2Cl8]4- is helder rood.
Verbindingen van hoofdgroepelementen hebben gevulde π-orbitalen en lege π*-orbitalen. Ze
zijn dikwijls kleurloos (bijv. N2 en CO), omdat het eenrgieverschil tussen deze orbitalen
overeenkomt met de energie van ultraviolette straling.
Bijkomende elektronen bezetten de δ*-orbitalen en reduceren de bindingsorde (herinner:
de bindingsorde is het verschil tussen het aantal elektronen in bindende moleculaire orbitalen
en het aantal elektronen in antibindende moleculaire orbitalen). Bijvoorbeeld, [Os2Cl8]2- is een
osmium(II)deeltje met een totaal van 10 d-elektronen. Dit complex heeft een drievoudige
binding. De δ-bindingsorde is nul. Omwille van de afwezigheid van een δ-binding, heeft dit
complex in tegenstelling tot [Re2Cl8]2- geen geëclipseerde conformatie, maar een gestaggerde
conformatie (D4d symmetrie), zoals verwacht op basis van VSEPR-theorie. Ook minder dan
acht valentie-elektronen geven een bindingsorde van minder dan vier.
Metaal-metaalbindingen hebben een zeer uitgesproken effect op de bindingsafstanden.
Een manier om de verkorting van de interatoomafstanden door meervoudige bindingen te
beschrijven is door de bindingsafstanden in meervoudige bindingen te vergelijken met de
bindingsafstanden in enkelvoudige bindingen. De verhouding van deze afstanden wordt soms
de formele verkortingsverhouding genoemd. De verhoudingen bedragen 0,783 voor C≡C,
0,786 voor N≡N, 0,767 voor Cr ≡ Cr, 0,807 voor Mo ≡ Mo en 0,848 voor Re ≡ Re. De
verhoudingen voor de viervoudig gebonden chroomcomplexen zijn de kleinste die voor om
het even welk type van verbindingen zijn gevonden. De variatie in bindingsafstanden van
viervoudige bindingen is vrij groot. Zo variëren Mo ≡ Mo bindingen tussen 203,7 en 230,2
pm. Daarentegen heeft het verwijderen van een elektron uit een δ*-orbitaal door oxidatie
slechts een zeer beperkt effect op de bindingslengte. Bijvoorbeeld de bindingslengte van de
Re ≡ Re binding bedraagt 224,1 pm in Re2Cl4(PMe2Ph)4 en 221,8 pm in [Re2Cl4(PMe2Ph)4]+.
Een mogelijke verklaring voor het geringe effect op de bindingslengte is dat de d-orbitalen
samentrekken met toenemende oxidatietoestand van het metaal. Deze samentrekking zorgt dat
de overlap van de d-orbitalen om π-bindingen aan te gaan minder efficiënt is. Bijgevolg
worden de π-interacties zwakker als de δ*-elektronen worden verwijderd. De twee factoren
(toename in bindingsorde en toename in de oxidatietoestand van renium) heffen elkaar bijna
volledig op.
69
Door een analogie te maken met de dubbele en drievoudige bindingen in de organische
chemie, zou men kunnen besluiten dat de viervoudige bindingen tussen transitiemetaalatomen
zeer sterk zijn. Men heeft echter gevonden dat de de bindingsenergie van de viervoudige
binding tussen 40 en 100 kJ/mol gelegen is voor 3d-elementen (bijv. Cr) en tussen 250 en 450
kJ/mol voor de 4d- en 5d-elementen (bijv. Mo, Re). Vergelijkt men dit met de
bindingsenergie van 346 kJ/mol voor een C-C binding, 610 kJ/mol voor een C=C binding en
836 kJ/mol voor een C≡C binding, dan is het duidelijk dat in veel gevallen de viervoudige
metaal-metaalbinding nog zwakker is dan een enkelvoudige koolstof-koolstofbinding. Deze
zwakte van een viervoudige metaal-metaalbinding lijkt paradoxaal, maar men kan de metaalmetaalbindingen moeilijk vergelijken met bindingen tussen kleine atomen (zoals koolstof)
omdat deze laatste voor meervoudige bindingen gebruik maken van p-orbitalen en de overlap
tussen p-orbitalen veel groter is dan de overlap tussen d-orbitalen, waardoor de overlap van porbitalen tot sterkere bindingen leidt.
2.4 Polymetallische clusters
De vorming van metaalclusters kan verklaard worden door het streven van metaalatomen
naar een edelgasconfiguratie. Bindingen tussen atomen dienen om te compenseren voor het
gebrek aan elektronen dat vereist is om de volgende edelgasconfiguratie in het periodiek
systeem te bereiken. Twee atomen winnen elk één elektron in hun valentieschil door een
elektronenpaar te delen. Als twee elektronen twee atomen verbindingen, spreekt men over
twee-elektron twee-centra bindingen, afgekort tot (2c2e)-bindingen. Als voor een element het
aantal beschikbare partneratomen van een verschillend element niet voldoende is om de
valentieschil te vullen, dan combineren atomen van eenzelfde element zich met elkaar zoals
het geval is in een groot aantal organische verbindingen. Voor de meer elektropositieve
elementen (de metalen) die in de eerste plaats al zelf over een kleiner aantal valentieelektronen
beschikken
en
die
bovendien
elektronen
moeten
afstaan
aan
meer
elektronegatievere partners, is het aantal beschikbare elektronen vrij klein. Deze verbindingen
kunnen op twee manieren elektronen bijkrijgen: (1) door complexvorming met liganden ter
vorming van coördinatieverbindingen; (2) door met hun eigen atomen bindingen te vormen,
ter vorming van (metaal)clusters. Als door de clustervorming voldoende elektronen
beschikbaar zijn zodat er een elektronenpaar is voor elke verbindingslijn tussen twee atomen,
dan kan elk van deze lijnen als een (2c2e)-binding beschouwd worden, juist zoals in de
gewone
valentiebindingsformules
(Lewis-structuren).
Dit
type
van
clusters
wordt
elektronprecieze clusters genoemd. Hoofdgroepelementen met minder dan vier valentieelektronen hebben niet voldoende elektronen om met behulp van (2c2e)-bindingen hun
70
gebrek aan elektronen te compenseren. Hun atomen vormen multicentra-bindingen. Het type
van cluster dat wordt gevormd noemt men een elektrondeficiënte cluster. In een drie-centra
twee-elektron binding (3c2e) delen drie atomen een elektronenpaar. Een elektronenpaar kan
zelfs tussen meer dan drie atomen worden verdeeld. Naarmate dat het aantal atomen dat een
elektronpaar moet delen echter toeneemt, wordt elk atoom minder stevig gebonden. Het
elektronenpaar in een (3c2e)-binding is grotendeels gelokaliseerd in het centrum van de
driehoek die door de drie atomen wordt gevormd. De positie van elektronenparen die meer
dan drie atomen met elkaar verbinden kan niet eenvoudig grafisch worden weergegeven. Bij
vele clusters vormt het metaalskelet een goed gekarakteriseerde polyeder. Zulke clusters
noemt men closo-clusters. Als de polyeder enkel uit driehoeken bestaat, spreekt men over een
deltaëder. Voorbeelden zijn de tetraëder, de trigonale bipiramide en de octaëder.
Tabel 2.1. Aantal elektronen geleverd door liganden aan metaalatomen in complexen, als de
metaalatomen als ongeladen worden geschouwd.
ligand
μ*
elektronen
ligand
μ*
elektronen
H
μ1
1
NR3
μ1
2
H
μ2
1
NCR
μ1
2
H
μ3
1
NO
μ1
3
CO
μ1
2
PR3
μ1
2
CO
μ2
2
OR
μ1
1
CS
μ1
2
OR
μ2
3
CR2
μ1
2
OR2
μ1
2
η2-C2R4
μ1
2
O, S, Se, Te
μ1
0
η2-C2R2
μ1
2
O, S, Se, Te
μ2
2
η5-C5R5
μ1
5
O, S, Se, Te
μ3
4
η -C6R6
μ1
6
O, S
int
6
C
int
4
F, Cl, Br, I
μ1
1
SiR3
μ1
2
F, Cl, Br, I
μ2
3
N, P
int
5
Cl, Br, I
μ3
5
6
*: μ1 = eindstandig ligand; μ2 = brugvormend ligand tussen twee atomen; μ3 = brugvormend
ligand tussen drie atomen; int = interstitieel atoom binnenin een metaalcluster.
Bij de elektronenrijke transitiemetalen (vanaf groep 6 in het periodiek systeem), komen
een groot aantal metaalclusters voor waarbij er voldoende elektronen aanwezig zijn om voor
71
elke zijde van de polyeder een elektronenpaar ter beschikking te stellen. Bovendien verkrijgt
elke metaalatoom in de cluster elektronen van de liganden zodat elk metaalatoom naar 18
valentie-elektronen streeft. De eenvoudigste manier om een elektronentelling uit te voeren is
te starten met de ongeladen metaalatomen en de ongeladen liganden. Liganden zoals NH3,
PR3 of CO stellen twee elektronen ter beschikking. Niet-brugvormende halogeenatomen,
waterstofatomen en groepen zoals SiR3 stellen één elektron ter beschikking. Bij
halogeenatomen is een alternatieve benadering te beschouwen dat we een Hal--ligand hebben
dat twee elektronen levert, maar dat voordien één elektron van een metaalion heeft verkregen.
Een μ2-brugvormend halogeenatoom levert drie elektronen (één zoals voordien en één vrij
elektronenpaar) en een μ3-brugvormend halogeenatoom levert vijf elektronen. In Tabel 2.1
wordt weergegeven hoeveel elektronen er door een welbepaald ligand aan een metaalatoom
worden geleverd. Uiteindelijk moet ook de lading aan de elektronentelling in rekening
gebracht worden.
De elektronen geleverd door de liganden en door de valentie-elektronen van de n
metaalatomen van een Mn-cluster geeft het totale elektronenaantal g. Het aantal M-M
bindingen (polyederzijden) b wordt dan gegeven door:
Cluster van hoofgroepelementen:
b = ½(8n – g)
Cluster van transitiemetaalelementen:
b = ½(18n – g)
Deze regel kan op een metaalcluster toegepast worden als het aantal elektronen voldoende is
om een elektronenpaar toe te kennen aan elke zijde van de polyeder die door de metaalatomen
wordt gevormd, en als de octetregel wordt gevolgd voor een hoofdgroepelement of de 18elektronenregel voor een transitiemetaal. Het aantal metaal-metaalbindingen b dat op deze
manier worden berekend is een ondergrens: het aantal polyederzijden in de cluster kan groter
of gelijk zijn aan b, maar nooit kleiner. Het aantal zijden is gelijk aan b als het over een
elektronprecieze cluster gaat.
Aangezien een atoom M één elektron per M-M-binding verkrijgt, kan de berekening ook
op de volgende manier worden uitgevoerd: het totale aantal valentie-elektronen g van de
cluster moet gelijk zijn aan:
Cluster van hoofgroepelementen:
g = 7n1 + 6n2 + 5n3 + 4n4
Cluster van transitiemetaalelementen:
g = 17n1 + 16n2 + 15n3 + 14n4
72
Hierbij zijn n1, n2, n3 en n4 het aantal polyederhoekpunten waarbij 1, 2, 3 of 4 polyëderzijden
(M-M-bindingen) elkaar raken. Polyeders met vijf of meer zijden per hoekpunt zijn in het
algemeen niet elektron-precies, waardoor er geen getallen n5, n6, ... in de bovenstaande
vergelijkingen voorkomen. De verwachte waarden voor de valentie-elektronen zijn in Tabel
2.2 weergeven. Er worden in deze tabel geen waarden gegeven voor de octaëder voor
hoofdgroepelementen, omdat octaëdrische clusters van hoofdgroepelementen zoals Tl68- niet
elektron-precies zijn.
Tabel 2.2. Aantal valentie-elektronen in metaalclusters voor verschillende polyeders
polyeder
hoofdgroepelementen
transitiemetaalelementen
driehoek
18
48
tetraëder
20
60
trigonale bipiramide
22
72
octaëder
--
84
trigonaal prisma
30
90
kubus
40
120
Voorbeelden van de toepassing van elektrontelregels in metaalclusters zijn hieronder
weergeven:
Os3(CO)12
3 Os
3 × 8 = 24
12 CO
12 × 2 = 24
⎯⎯⎯⎯⎯
g=
48
= 16 n2
b = ½ (18 × 3 – 48) = 3
Ir4(CO)12
4 Ir
4 × 9 = 36
12 CO
12 × 2 = 24
⎯⎯⎯⎯⎯
g=
60
= 15 n3
b = ½ (18 × 4 – 60) = 6
73
[Os6(CO)18P]6 Os
6 × 8 = 48
18 CO
18 × 2 = 36
P
5
lading
1
⎯⎯⎯⎯⎯
g=
90
= 15 n3
b = ½ (18 × 6 – 90) = 9
[Mo6Cl14]26 Mo
6 × 6 = 36
8 μ3-Cl
8 × 5 = 40
6 μ1-Cl
6×1= 6
lading
2
⎯⎯
g=
84
= 14 n4
b = ½ (18 × 6 – 84) = 12
De vaste stof MoCl2 kan gesynthetiseerd worden door MoCl5 in een afgesloten glasbuis te
laten reageren met een mengsel van gesmolten NaAlCl4 en AlCl3, in de aanwezigheid van
aluminiummetaal als reducerend reagens:
NaAlCl4 / AlCl3 ( l ), 200 °C
MoCl5(s) + Al(s) ⎯⎯
⎯⎯⎯⎯⎯
⎯→ MoCl2(s) + AlCl3(l)
Deze verbinding is onder milde omstandigheden tegen oxidatie bestand. Wanneer ze wordt
behandeld met waterstofchloride, wordt de anionische metaalcluster [Mo6Cl14]2- gevormd.
Deze bestaat uit een Mo6-cluster die in een Cl8-cluster is ingeschreven. Elk van de acht
chlooratomen van de kubus is geplaatst bovenop één van de octaëdervlakken en is met drie
molybdeenatomen gecoördineerd (Figuur 2.8). In deze formule-eenheid [Mo6Cl8]4+ heeft elk
molybdeenatoom twee elektronen te kort om 18 valentie-elektronen te hebben. Deze worden
geleverd door de zes chloride-ionen die verbonden zijn met de hoekpunten van de octaëder.
Deze chloride-ionen kunnen vervangen worden door andere halogenide-ionen, door alkoxiden
of door fosfines. Een analoge reeks van wolframclusters is ook bekend. MoCl2 zelf bestaat uit
Mo6-octaëders die in een kubus van zes chlooratomen zijn ingeschreven. In tegenstelling tot
[Mo6Cl14]2-, zijn er per cluster slechts vier chloride-ionen nodig om het te kort aan elektronen
in de [Mo6Cl8]4+-eenheid te compenseren. Elk hoekpunt van de octaëder draag weliswaar nog
74
een chloride-ion, maar vier van deze chloride-ionen treden op als brugvormende ligand en
wordt met een andere [Mo6Cl8]4+-eenheid gedeeld. Hierdoor heeft MoCl2 (dat eigenlijk beter
als Mo6Cl12 zou geschreven worden) een laagvormige structuur (Figuur 2.9).
Figuur 2.8. Twee representaties van de [Mo6Cl8]4+-cluster (boven) en de structuur
van het [Mo6Cl14]2--ion (onder).
Figuur 2.9. Reeks van Mo6Cl8-clusters die via chlooratomen tot een
laag verbonden zijn in Mo6Cl12 (MoCl2).
75
Als er niet voldoende elektronen beschikbaar zijn om alle zijden van de clusterpolyeder,
zijn (3c2e)-bindingen op de driehoeksvlakken van een polyeder een oplossingen om te
compenseren voor het gebrek aan elektronen. Deze oplossing is enkel mogelijk voor
deltaëders (d.w.z. polyeders die uit driehoeksvlakken zijn opgebouwd) waarbij niet meer dan
vier zijden (of vlakken) op een hoekpunt samenkomen, zoals de tetraëder, de trigonale
bipiramide en de octaëder. Bijvoorbeeld, de binding in B4Cl4 kan geïnterpreteerd worden op
de volgende wijze: elk booratoom neemt deel aan vier bindingen, zijnde één (2c2e)-binding
B-Cl en drie (3c2e)-bindingen op de vlakken van de B4-tetraëder. Op deze manier verkrijgt
elk booratoom een octetconfiguratie. Acht van de valentie-elektronen nemen deel aan de
vorming van meercentrabindingen, terwijl de andere acht nodig zijn voor de B-Cl bindingen.
Men veronderstelt dat in het [Nb6Cl18]4--ion voor de octaëdrische Nb6-cluster acht (3c2e)bindingen voorkomen op de acht octaëdervlakken. Bovenop elke zijde van de octaëder
bevindt zich een chlooratoom dat twee niobiumatomen verbindt. Aangezien een octaëder
twaalf zijden heeft, vormen deze twaalf chlooratomen een [Nb6Cl12]2+-eenheid. De
overblijvende zes chooratomen zijn eindstandig verbonden met de hoekpunten van de
octaëder. De telling van de valentie-elektronen is in het [Nb6Cl18]4--ion is:
6 Nb
6×5=
30
12 μ2-Cl
12 × 3 =
36
6 μ1-Cl
6×1=
6
lading
4
⎯⎯
76
Van deze 76 elektronen zijn 12 elektronen nodig voor de bindingen met de eindstandige
chlooratomen (μ1-Cl). Vier elektronen zijn vereist voor elk chloor-atoom bovenop een
octaëderzijde, in totaal 4 × 12 = 48. 76 – 12 – 48 = 16 elektronen blijven over voor de Nb6octaëder, d.w.z. juist één elektronenpaar per vlak van de octaëder. De situatie voor elk Nbatoom is dezelfde als in het [Mo6Cl14]2--ion: het metaalatoom is omringd door vijf
chlooratomen en is betrokken bij vier metaal-metaalbindingen. De positie van de MCl5eenheid is echter in beide clusters verschillend: de chlooratomen bovenop de vlakken van de
Mo6-octaëder worden chlooratomen bovenop de zijden van de Nb6-octaëder. In beide
gevallen is het aantal valentie-elektronen per metaalatoom 18. In Figuur 2.10 worden de
structuur van de [Nb6Cl12]2+ en van de [Nb6Cl18]4- clusters weergegeven. In de verbinding
Nb6Cl14 zijn de [Nb6Cl12]2+-clusters via brugvormende chloride-atomen aan elkaar geknoopt,
zoals in Mo6Cl12.
76
Figuur 2.10. Structuur van de [Nb6Cl12]2+-cluster (links) en van de
[Nb6Cl18]4--cluster (rechts).
Clusters van metalen die slechts enkele valentie-elektronen hebben kunnen hun gebrek
aan elektronen ongedaan maken door atomen op te sluiten in de holte die door de
clusterpolyeder wordt gevormd (Figuur 2.11). Dit zijn metaalclusters met interstitiële atomen.
Deze optie wordt vooral door octaëdrische clusters gevolgd. Het interstitieel atoom stelt al
zijn valentie-elektronen ter beschikking van de elektronenbalans. Als interstitiële atomen
komen niet-metalen zoals H, B, C, N en Si voor, evenals metalen zoals Be, Al, Mn, Fe, Co en
Ir. Transitiemetalen van groepen 3 en 4 vormen vele octaëdrische clusters die isostructureel
zijn met deze van minder elektrondeficiënte elementen van de volgende groepen, maar ze
bevatten extra atomen in hun centra. Vertrekkende van het hierboven vermelde Nb6Cl14,
kunnen we de niobiumatomen door zirconiumatomen vervangen. Het aantal beschikbare
elektronen wordt dan met zes verminderd. Dit verlies kan gedeeltelijk worden gecompenseerd
door een koolstofatoom binnen in een Zr6-octaëder te plaatsen. Alhoewel Zr6CCl14 te weinig
elektronen heeft om aan de 18-elektronenregel te voldoen, is deze cluster toch stabiel door
een verschil in binding. Het elektronegatievere atoom in het centrum van de cluster trekt de
elektronenwolk van de cluster naar binnen, waardoor de Zr-Zr-bindingen gedeeltelijk worden
verzwakt, ten gunste van sterkere interacties van de Zr-atomen met het C-atoom. Metaalmetaalbindingen worden daarentegen versterkt als het interstitieel atoom een metaalatoom is.
Zo bestaat Nb6F15 uit [Nb6F12]-clusters van hetzelfde type als de [Nb6Cl12]2+-eenheid. De
[Nb6F12]-eenheden zijn via alle zes hoekpunten van de octaëder aan elkaar verbonden via
brugvormende fluoratomen, ter vorming van een netwerk. Th6FeBr15 heeft eenzelfde type van
structuur, maar met een bijkomend ijzeratoom in het centrum van de octaëder. Nb6F15 heeft
één elektron te weinig om te voldoen aan de vereiste elektronen voor acht (3c2e)-bindingen.
In Th6Br15 ontbreken zes bijkomende elektronen. Het interstitieel Fe-atoom (d 8 ) levert deze
zeven elektronen. Het achtste elektron blijft bij het Fe-atoom.
77
Figuur 2.11. Cluster-eenheid met een interstitieel atoom in verbindingen
zoals Zr6CCl14 en Th6FeBr15.
2.5 Zintl-ionen
Het is reeds vanaf het begin van de 20ste eeuw bekend dat posttransitiemetalen zoals tin,
lood, antimoon en bismut oplossen in vloeibare ammoniak in de aanwezigheid van
alkalimetalen ter vorming van diepgekleurde oplossingen. In de jaren 1930 vond de Duitse
chemicus Eduard Zintl dat er hierbij polyatomische clusters zoals Pb52+, Pb74-, Pb94-, Sn94-,
Sb73- of Bi95+ werden gevormd, die later ter zijner ere Zintl-ionen (Zintl-kationen en Zintlanionen) werden genoemd (Figuur 2.12). Aangezien de Zintl-ionen geen liganden dragen,
worden ze soms ook “naakte clusters” genoemd. De elektronentelregels die voor boor- en
carboraanclusters worden gebruikt (regels van Wade) zijn ook toepasbaar op de Zintl-ionen,
vooral dan op de Zintl-anionen. Beschouw als voorbeeld de elektronentelling in Pb52-. Het
aantal valentie-elektronen bedraagt 5 × 4 = 20 van de loodatomen en twee bijkomende
elektronen van de lading, dus 22 elektronen (of 11 elektronenparen) in totaal. Van deze 11
elektronenparen zijn 5 paren (1 paar per Pb-atoom) niet beschikbaar voor binding, omdat dit
elektronenpaar zoals het elektronpaar van de B-H-eenheid in boranen weggericht is van het
skelet dat door de 5 Pb-atomen wordt gevormd. Het totaal aantal elektronenparen dat
beschikbaar is voor vorming van het metaalskelet is 11 – 5 = 6. Dit getal is in
overeenstemming met de elektronentelling die verwacht worden voor een closo-cluster
bestaande uit vijf atomen (n = 5).
78
Figuur 2.12. Representatieve voorbeelden van Zintl-ionen:
(a) Pb55-, (b) Sn94-, (c) Bi95+, (d) Te64+.
79
Hoofdstuk 3:
Karakteristieke structuren van vaste stoffen
Men kan kristallen in twee grote klassen onderverdelen: (1) moleculaire kristallen en (2)
niet-moleculaire
kristallen.
De
niet-moleculaire
kristallen
kunnen
verder
worden
onderverdeeld in (a) metallische kristallen, (b) covalente kristallen en (c) ionische kristallen,
afhankelijk van de dominante bindingskracht waaraan het kristal zijn coherentie dankt. Het
type van chemische binding in kristallen is echter bijna nooit volledig metallisch, covalent of
ionische van aard., en is meestal een hybride van deze uitersten.
3.1 Moleculaire kristallen
Moleculaire kristallen bestaan uit een regelmatige stapeling van moleculen. De
(grotendeels) covalente binding tussen de atomen van het molecule is veel sterker dan de
binding tussen de moleculen onderling. De chemische binding tussen de moleculen onderling
in een moleculair kristal zijn vaak slechts zwakke van der Waalskrachten. Ook
waterstofbruggen of π-π-interacties komen regelmatig voor.
Als gevolg van deze zwakke cohesiekrachten zijn deze kristallen vaak zacht en weinig
stevig: ze kunnen gemakkelijk worden gekrast of gebroken. Als de temperatuur stijgt en de
thermische vibraties van de moleculen in het kristal steeds toenemen, zal een moleculair
kristal
meetbaar
in
volume
uitzetten;
een
moleculair
kristal
heeft
een
grote
uitzettingscoëfficiënt. Dit komt omdat bij verhoogde temperatuur de zwakke van der
Waalskrachten niet in staat zijn om de moleculen door de toegenomen thermische beweging
bij elkaar te houden. Een molecule aan het oppervlak van een moleculair kristal kan zodanig
bewegen dat het de van der Waalsbindingen met de omringende moleculen verbreekt en uit
het kristal ontsnapt. Als een moleculair kristal onder vacuum in een afgesloten container
wordt verhit, dan zorgen de moleculen die uit het kristal zijn ontsnapt voor een meetbare
dampspanning. De dampspanning is relatief hoog voor moleculaire kristallen en neemt toe
met toenemende temperatuur. Als de temperatuur blijft toenemen worden de vibraties
krachtig genoeg om alle bindingen tussen de moleculen te verbreken, niet alleen aan het
oppervlak, maar ook diep in het kristal. Op dat ogenblik smelt het kristal. De regelmatige
stapeling van moleculen gaat verloren en de moleculaire mobiliteit neemt toe. Omdat
moleculaire kristallen hun bestaan danken aan de cohesie door zwakke van der
Waalskrachten, smelten moleculaire kristallen bij relatief lage temperaturen. Ze zijn
bovendien slechte geleiders van elektriciteit (isolatoren).
80
Alhoewel moleculaire kristallen veel voorkomen bij organische verbindingen en
coördinatieverbindingen, zijn ze voor anorganische verbindingen eerder zeldzaam. Mineralen
die als moleculaire kristallen voorkomen zijn zwavel (S8), realgar (As4S4), calomel (Hg2Cl2),
arsenoliet (As4O6), senarmontiet (Sb4O6) en ijs (H2O). In figuur 3.1. wordt de kristalstructuur
van zwavel getoond.
Figuur 3.1. Kristalstructuur van de orthorhombische polymorf van zwavel. De S8-moleculen
zijn duidelijk te zien.
3.2 Bolstapelingen
Vele kristalstructuren kunnen beschouwd worden als regelmatige stapelingen van atomen
of ionen. Vandaar dat we eerst de meest voorkomende typen van bolstapelingen beschouwen.
Hierbij wordt bekeken op welke manieren harde (niet-vervormbare) bollen zoals biljartballen
op de meest efficiënte wijze ordentelijk kunnen worden gestapeld, d.w.z. op welke wijze de
beschikbare ruimte optimaal kan worden opgevuld. Als men een aantal bollen van gelijke
grootte op de bodem van een rechhoekige doos plaatst, met de beperking dat de ordening van
de bollen regelmatig moet zijn, is de meest efficiënte manier om de bodem van de doos te
bedekken deze die in Figuur 3.2 wordt weergegeven. Zulk een ordening is een dichtste
81
stapeling. De bollen die zich niet aan de zijkanten van het geheel bevinden, zijn in de laag in
contact met zes andere bollen, waardoor er in de laag bollen een regelmatig hexagonaal
patroon aanwezig is. In Figuur 3.3 wordt een deel van deze dichtste stapeling van bollen
getoond. Tussen de bollen kan men holten herkennen. Men kan een twee laag bollen op de
onderste laag plaatsen door door deze bijkomende bollen in de holten van de eerste laag te
plaatsen. Als men de bollen op een zodanige wijze in de twee laag plaatst dat er opnieuw een
dichtste stapeling in deze laag wordt gevormd, dat is slechts de helft van de holten van de
eerste laag met nieuwe bollen gevuld. Het aantal holten in de laag is immers dubbel zo veel
als het aantal bollen in die laag.
Figuur 3.2. Als de bodem van een rechthoekige doos wordt bedekt met bollen van gelijke
grootte, met de beperking dat de laag moet geordend zijn, dan is de meest efficiënte plaatsing
van bollen een dichtste stapeling. In stippellijn wordt de hexagonale herhalingseenheid
weergegeven.
Figuur 3.3. Opbouw van een hexagonale dichtste bolstapeling (ABABAB) en een kubische
dichtste bolstapeling (ABCABC).
82
Als men deze stapeling van twee lagen bollen van bovenaf bekijkt, zijn er twee typen van
holten zichtbaar. Holten van type A (A-holten) liggen direct boven een bol in de onderste
laag. Holten van type C (C-holten) liggen juist boven een holte in de onderste laag. Het
gevolg is dat wanneer men een derde laag van bollen toevoegt, er twee mogelijke dichtste
bolstapelingen mogelijk zijn. Als men de bollen van de derde laag in de holten boven de
bollen van de eerste laag plaatst, is de ordening van de bollen in de derde laag identiek aan
deze van de eerste laag. Als men de bollen van de derde laag echter plaatst boven de nog lege
holten van de eerste laag, krijgt men een derde mogelijke laag. In dit geval zal de vierde laag
dezelfde stapeling hebben als de eerste laag. Men onderscheidt op deze manier twee
verschillende
dichtste
bolstapelingen,
namelijk
één
met
twee
repeterende
lagen,
ABABABAB... en één met drie repeterende lagen, ABCABCABC.... De ABABAB-stapeling
wordt de hexagonale dichtste bolstapeling (hdb) genoemd en de ABCABC-stapeling wordt de
kubische dichtste bolstapeling genoemd (kdb). Men vindt in de literatuur ook de afkortingen
hcp (hexagonal close packing) en ccp (cubic close packing) terug. In elk van de twee dichtste
bolstapelingen maakt elke bol contact met 12 andere bollen (elke bol is omringd door 12
andere bollen). Men zegt dat elke bol 12 naaste buren heeft, dat deze bol coördinatiegetal 12
heeft of dat deze bol 12-voudig is gecoördineerd. In Figuur 3.4 wordt getoond hoe dit
coördinatiegetal wordt verkregen. De coördinatiepolyeder voor een kubische dichtste
bolstapeling is een kuboctaëder en deze voor een hexagonale dichtste bolstapeling is een antikuboctaëder. Een kuboctaëder kan beschouwd worden als een getrunceerde kubus of een
getrunceerde octaëder. De anti-kuboctaëder wordt uit de kuboctaëder verkregen door de twee
tenoverstaande driehoeksvlakken over 30° te draaien. Er bestaan naast de hexagonale en
kubische dichtste bolstapelingen, nog andere dichtste bolstapelingen, maar met een
ingewikkelder stapelingspatroon. Deze komen minder frequent voor maar worden voor
sommige lanthaniden waargenomen. Bijvoorbeeld, de stapelingsvolgorde van de lagen is
....ABAC.... in La, Pr, Nd, Pm en ....ABACACBCB... in Sm.
83
Figuur 3.4. Coördinatiesfeer van bol in een (a) hexagonale dichtste bolstapeling ABA en (b)
in een kubische dichtste bolstapeling ABC. De coördinatiepolyeder voor de hexagonale
dichtste bolstapeling is een anti-kuboctaëder en deze voor een kubische dichtste bolstapeling
is een kuboctaëder. In beide gevallen is de centrale bol in het middenvlak omgeven door zes
andere bollen, met nog drie bollen erboven en eronder.
De “eenheidscel” is een fundamenteel begrip in de vaste-stofchemie. Het is de kleinste
repeterende eenheid van de structuur die alle informatie bevat om op een ondubbelzinnige
manier een oneindig kristalrooster te construeren. In Figuur 3.4 worden de eenheidscellen
voor de hexagonale en kubische dichtste bolstapelingen weergegeven. Een kubische dichtste
bolstapeling wordt ook een kubische vlakkengecenterde structuur (kvg) genoemd (fcc, facecentered cubic). Uit Figuur 3.5 is duidelijk waarom men deze benaming gebruikt. De
eenheidscel voor een hexagonale dichtste bolstapeling bestaan uit delen van drie ABA lagen,
waardoor de relatie tussen de eenheidscel en de ABABAB-volgorde gemakkelijk te
herkennen is. Het is moeilijker om de ABCABCABC volgorde te herkennen in de eenheidscel
van de kubische bolstapeling. Dit is te wijten aan het feit dat de ABCABCABC lagen niet
parallel zijn met het grondvlak van de eenheidscel, maar wel met een diagonaalvlak door de
kubus. In Figuur 3.6. worden de kubische en hexagonale dichtste bolstapelingen nog eens
duidelijk met elkaar vergeleken.
84
Figuur 3.5. Eenheidscellen van een kubische dichtst gestapeld rooster (links) en van een
hexagonaal dichtst gestapeld rooster (rechts).
Figuur 3.6. Vergelijking tussen de kubische dichtste bolstapeling (boven) en de hexagonale
dichtste bolstapeling (onder). Links staat telkens de coördinatiepolyeder, in het midden de
eenheidscel en rechts het stapelingspatroon.
Dichtste bolstapelingen bevatten tetraëdrische en octaëdrische holten. Figuur 3.7 toont
twee verschillende voorstellingen van twee lagen van bollen in een dichtste bolstapeling.
Figuur 3.7a is de ruimtevullende voorstelling, terwijl in Figuur 3.7b de grootte van de bollen
85
werd gereduceerd zodat de verbindingslijnen tussen de bollen duidelijk worden (“bol-enstaaf”-diagram). Deze laatste voorstelling laat zien dat de bollen gelegen zijn op de
hoekpunten van tetraëders of octaëders. De bollen stapelen zich bijgevolg zo dat er
octaëdrische en tetraëdrische holten tussen hen zijn. Per bol is er één octaëdrische holte in een
dichtste bolstapeling. Het aantal tetraëdrische holten is dubbel zo groot als het aantal
octaëdrische holten. De octaëdrische holten zijn groter dan de tetraëdrische holten. Terwijl er
in een tetraëdrische holten plaats is voor een bol met een straal die ongeveer 0,23 keer deze
van de dichtst gestapelde bollen is, past een bol met een straal die ongeveer 0,41 keer deze
van de dichtste gestapelde bollen is in een octaëdrische holte.
Figuur 3.7. Twee lagen van dichtst-gestapelde atomen. (a) Ruimtevullende voorstelling; (b)
Bol-en-staaf-voorstelling waarin de octaëdrische en tetraëdrische holten worden getoond.
Bollen worden niet altijd even efficiënt als in een dichtste bolstapeling gestapeld. Men
kent geordende stapelingen van bollen, waarbij de ruimtevulling minder bedraagt dan de 74%
die bij de dichtste bolstapeling voorkomt. De bollen kunnen zodanig geplaatst worden dat de
eenheidscel een eenvoudige kubus is, waardoor men een primitief kubisch rooster krijgt. In
het kristalrooster is het coördinatiegetal van elke bol dan 6. De holte in elke kubus is niet
groot genoeg om plaats te bieden aan een bol van gelijke grootte. Als de acht bollen van de
kubische eenheidscel echter een beetje uit elkaar worden getrokken, is er wel voldoende
ruimte om een bijkomende bol in de holte te plaatsen. Het resultaat is een kubisch ruimtelijke
gecenterde structuur (krg), waarbij het coördinatiegetal van elke bol 8 is (bcc, body-centered
cubic) (Figuur 3.8). Alhoewel er slechts 8 naaste buren zijn op een afstand x (vergeleken met
12 in de kvg-structuur), zijn er 6 bijkomende buren op de afstand 1,15x.
86
Figuur 3.8 Eenheidscel van (a) een primitief kubisch rooster en (b) een kubisch ruimtelijk
gecenterd rooster.
Bij een dichtste bolstapeling wordt op de meest efficiënte wijze gebruik gemaakt van de
beschikbare ruimte. De fractie van de ruimte die door de bollen wordt bezet bedraagt π/(3√2)
= 0,7405 of 74,05%. De stapelingsefficiëntie (of stapelingsfactor) is hetzelfde voor alle typen
van dichtste bolstapelingen. Reeds in 1603 beweerde de beroemde astronoom Johannes
Kepler dat er geen stapeling van bollen mogelijk is die meer dan 74,05% van de ruimte vult.
Het suitende wiskundig bewijs werd pas in 1998 geleverd. Bij een kubisch ruimtelijk
gecenterde structuur is de stapelingsefficiëntie π√3/8 = 0,6802 of 68,02%. De
stapelingsefficiëntie voor een primitief kubisch rooster bedraagt slechts 52%.
3.3 Metalen en legeringen
3.3.1 Metaalbinding
Metalen en legeringen onderscheiden zich van alle andere vaste stoffen door hun grote
elektrische
en
thermische
geleidbaarheid,
door
hun
groot
reflectievermogen
en
ondoorlaatbaarheid voor zichtbaar licht, door hun uitstekende vervormbaarheid, taaiheid, ...
Ongeveer 75% van de elementen vertonen een metaalkarakter, evenals vele sulfiden,
seleniden, telluriden, arseniden, antimoniden en bismutiden van zware metalen.
Juist zoals in een ionisch of covalent kristalrooster streven de bouwstenen van een
metaalrooster ernaar om een edelgasconfiguratie te verkrijgen. Deze edelsgasconfiguratie kan
verkregen worden omdat de valentie-elektronen van de atomen gemakkelijk worden
afgegeven, waardoor een elektronengas ontstaat, waarin de positieve metaalionen zijn
ingebed. De chemische binding in metalen noemt men de metaalbinding. De Coulomb-
87
aantrekking tussen de positieve metaalionen en het negatief geladen elektronengas levert een
belangrijk gedeelte van de roosterenergie. De vrije beweeglijkheid van het elektronengas
onder thermische, elektrische of optische stimulatie, is verantwoordelijk voor de goede
thermische en elektrische geleibaarheid en voor het optisch gedrag. Eigenschappen zoals
vervormbaarheid staan in verband met de geometrisch eenvoudige kristalroosters.
Het metaalrooster wordt opgebouwd door bolvormige metaalatomen met een niet-gerichte
ladingsverdeling. Hierdoor zijn de kristalstructuren van vele metalen dichtste bolstapelingen
van metaalatomen. Door te stollen in een zo dichtst mogelijke gestapelde rangschikking is het
aantal bindingen per volume-eenheid zo groot mogelijk en dus de bindingsenergie per
volume-eenheid zo negatief mogelijk.
3.3.2 Structuur van metalen in de vaste toestand
Met de uitzondering van kwik zijn alle metalen vast bij 25 °C. De uitdrukking “vast bij
kamertemperatuur” is dubbelzinnig, omdat door de lage smeltpunten van cesium (28,4 °C) en
gallium (29,8 °C), deze metalen in sommige landen bij kamertemperatuur vloeibaar zijn. De
meeste metalen kristalliseren uit in structuren met ccp-, hcp- of bcc-roosters.
Een hexagonale dichtste bolstapeling wordt gevonden voor Be, Mg, Sc, Y, Ti, Zr, Hf, Tc,
Re, Ru, Os, Co, Ni, Tl. Een kubische dichtste bolstapeling vinden we voor Ca, Sr, Rh, Ir, Pd,
Pt, Ag, Au, Al, Pb, Th. Een kubisch ruimtelijk gecenterde structuur komt voor bij Li, Na, K,
Rb, Cs, Ba, V, Nb, Ta, Cr, Mo, W en Fe. Vele metalen vertonen echter allotropie (of
polymorfie) en vertonen meer dan één structuur afhankelijk van de voorwaarden voor druk en
temperatuur. De structuren van zink en cadmium ziijn gebaseerd op de hexagonale dichtste
bolstapeling, maar door verstoring van het kristalrooster heeft elk atoom slechts 6 naaste
buren (binnen eenzelfde laag van atomen) en nog 6 bijkomende buren op een grotere afstand.
Kwik kirstalliseert uit in een structuur met een primitieve kubische eenheidscel. Door
verstoring van het rooster heeft elk kwikatoom slechts 6 naaste buren. Zoals reeds hoger
vermeld komen bij verscheidene van de lanthaniden weliswaar dichtste bolstapelingen voor,
maar deze wijken af van de klassieke ABAB of ABCABC stapelingspatronen. Voor La, Pr,
Nd, Pm vinden we de ABAC-sequentie. Deze wordt ook wel een dubbele hexagonale dichtste
bolstapeling genoemd. Voor Sm vinden we de ABACACBCB-sequentie. De kristalstructuur
van Gd, Tb, Dy, Ho, Tm is een gewone hexagonale dichtste bolstapeling. Voor Ce en Yb
hebben een kubische dichtste bolstapeling en Eu vormt een kubisch ruimtelijk centerde
structuur.
Een aantal metalen vormt een kristalstructuur die niet op de ccp-, hcp- of bcc-roosters zijn
gebaseerd: Ga, Sn, Bi, Po, Mn, Pa, U, Np en Pu. Gallium heeft een vrij ongewone structuur
88
waarbij elk galliumatoom een coördinatiegetal 1+6: één van de 7 omringende galliumatomen
is beduidend dichter dan de andere (1× 244 pm en 6× 270-279 pm). Men heeft dit
geïnterpreteerd als zijnde een metaalstructuur die niet uit enkelvoudige atomen bestaat, maar
uit Ga-Ga paren met een covalente binding. Het zeer lage smeltpunt van gallium (29,8 °C)
duidt aan dat deze structuur niet erg stabiel is. Er lijkt ook geen optimale structuur te bestaan
voor Mn, U, Np en Pu, omdat deze elementen een opmerkelijk groot aantal polymorfen met
speciale structuren vormen. Bijvoorbeeld, voor mangaan bevat de eenheidscel van α-Mn (de
polymorf die bij kamertemperatuur stabiel is) bevat niet minder dan 58 atomen en er komen
vier verschillende coördinatiepolyeders voor met coördinatiegetallen 12, 13 en 16. Een
primitief kubisch kristalrooster heeft men tot hiertoe slechts éénmaal waargenomen, namelijk
voor een onstabiele fase van polonium, zijnde α-Po dat beneden –76 °C stabiel is.
3.3.3 Polymorfie bij metalen
De metaalbinding heeft in tegenstelling tot de covalente binding weinig directionaliteit,
maar strekt zich in alle richtingen uit. Een metaalbinding kan zich vormen op elk raakpunt
tussen naburige metaalatomen. Dit leidt niet enkel tot kristalstructuren die op dichtste
bolstapelingen zijn gebaseerd, maar zorgt er ook voor dat polymorfie wijd verbreid is bij
metalen. Metalen kunnen verschillende kristalstructuren aannemen, afhankelijk van de
voorwaarden voor druk en temperatuur. Zo vertoont ijzer verscheidene vast-vast transities als
het wordt verhit en de atomen nemen een nieuwe stapelingsvorm aan.
De polymorfen van metalen worden meestal gelabeld met een Griekse letter α,β, γ, ...,
waarbij α wordt gebruikt om de polymorf aan te duiden die stabiel is bij de laagste
temperatuur, en β, γ, ... voor de polymorfen die stabiel zijn bij hogere temperatuur. Sommige
metalen keren bij hoge temperaturen terug naar een structuur die bij lage temperaturen stabiel
is. Een voorbeeld hiervan vinden we bij ijzer. α-Fe heeft een kubisch ruimtelijk gecenterde
structuur en is stabiel tot 906 °C. Tussen 906 °C en 1401 °C komt γ-Fe voor. Dit polymorf
heeft een kubische dichtste bolststapeling. Van 1401 °C tot het smeltpunt bij 1530 °C is α-Fe
terug stabiel. β-Fe vormt zich enkel bij hoge drukken en zijn structuur is een hexagonale
dichtste bolstapeling.
De polymorf van tin die bij kamertemperatuur stabiel is, is wit tin (β-Sn). Wit tin
ondergaat een omzetting tot grijs tin (α-Sn) bij temperaturen lager dan 13,2 °C, maar de
omzetting vereist meestal langere blootstelling aan veel lagere temperaturen Bij daling van de
temperatuur neemt de drang naar het ondergaan van de overgang nog verder toe, maar bij al te
lage temperaturen neemt de groeisnelheid van de nieuwe fase af. De optimale temperatuur
89
voor de overgang is daarom rond -48°C. Wit tin is een zilverwit sterk glanzend metaal, terwijl
grijs tin poedervormig is. De overgang van wit naar grijs tin begint meestal vanuit een beperkt
aantal groeicentra en breidt zich langzaam uit. Aan voorwerpen uit tin is dat te zien als een
aantal donkere plekken die blaasjes en oneffenheden veroorzaken. Vandaar dat de faseovergang ook met de term “tinpest” wordt aangeduid. Tinpest was in de middeleeuwen een
onbegrepen en gevreesd verschijnsel omdat het orgelpijpen in stof deed uiteenvallen. Het
verhaal gaat dat tijdens de dramatische Russische veldtocht van Napoleon in de winter van
1812 vele Franse soldaten van koude zijn omgekomen omdat bij de extreem lage
temperaturen alle knopen van hun soldatenuniform tot poeder uiteenvielen. Tinpest kan ook
overgaan van het ene tinnen voorwerp naar het andere. Grijs tin heeft een kubische structuur
en de kristalstructuur heeft dezelfde opbouw als deze van diamant. In de tetragonale structuur
van wit tin zijn de tinatomen door zes andere tinatomen omgeven in de vorm van een
verstoorde octaëder. Alhoewel de vier naaste buren van wit tin zich op een grotere afstand
bevinden dan de vier naaste buren van grijs tin (wat te verwachten is voor een polymorf die
bij hogere temperatuur stabiel is), heeft wit tin echter een grotere massadichtheid (7,285
g/cm3) dan grijs tin (5,769 g/cm3). De verklaring is dat bij wit tin de atomen in de tweede
coördinatiesfeer zich op dichtere afstand bevinden dan in grijs tin, waardoor wit tin een
compactere vaste stof dan grijs tin is. Een interessante vaststelling is dat wanneer tin in een
geconcentreerde waterstofchloride-oplossing oplost, wit tin tin(II)chloride vormt, terwijl grijs
tin tin(IV)chloride vormt. Dit toont aan hoe de kristalstructuur van een verbinding de
chemische eigenschappen kan beïnvloeden.
Het komt vaak voor dat de dichtste bolstapelingen thermodynamisch het gunstigst zijn bij
lage temperaturen, terwijl minder dicht gestapelde structuren bij hogere temperaturen
voorkomen. Dit verschijnsel kan ook door middel van de Regel van Goldschmidt worden
uitgedrukt: “Temperatuurstoename begunstigt structuren met lagere coördinatiegetallen”.
Dichtste bolstapelingen worden ook bij verhoging van de druk aangenomen. De kubisch
ruimtelijk gecenterde structuur komt vaak voor bij hogere temperaturen voor metalen die bij
lagere temperaturen een dichtste bolstapelingsstructuur hebben, omdat de toegenomen
amplitudes van vibraties in de hete vaste stof een minder dicht gepakte structuur vereisen.
Voor vele metalen, zoals calcium, titaan en mangaan, ligt de transitietemperatuur boven
kamertemperatuur.
Voor
andere
metalen,
zoals
lithium
en
natrium,
ligt
de
transitietemperatuur beneden kamertemperatuur. Men heeft empirisch waargenomen dat de
kubisch ruimtelijk gecenterde structuur begunstigd wordt door een klein aantal valentieelektronen. Deze waarneming suggereert dat er een dichte elektronenwolk noodzakelijk is om
de kationen in een dichtste bolstapeling samen te trekken. De alkalimetalen hebben niet
voldoende valentie-elektronen om een dichtste bolstapeling te verkrijgen.
90
3.3.4 Legeringen
Een legering is een mengsel van metalen dat bereid wordt door de gesmolten
componenten te mengen en vervolgens het mengsel af te koelen. Verschillende metalen
kunnen vaak met elkaar in de gesmolten toestand gemengd worden, waarbij er homogene
oplossingen worden gevormd. Door de smelt snel af te koelen wordt er een vaste oplossing
gevormd (zie hoofdstuk 4). In de wanordelijke legering die op deze manier wordt verkregen
zijn de atomen willekeurig in de vaste stof verdeeld. Wanneer de smelt traag wordt afgekoeld,
kunnen in sommige gevallen ook vaste oplossingen worden verkregen. Het komt echter vaker
voor dat er een afscheiding van meerdere fasen plaatsvindt. Dit kan op verschillende manieren
gebeuren:
(1) De metalen kristalliseren onafhankelijk van elkaar uit (volledige afscheiding).
(2) Twee typen van vaste oplossingen kristalliseren uit, een oplossing van metaal 1 in
metaal 2, en een oplossing van metaal 2 in metaal 1 (beperkte oplosbaarheid)
(3) Een legering met een welbepaalde samenstelling kristallieert uit (vorming van een
intermetallische verbinding). De samenstelling van de legering kan verschillend zijn
van de samenstelling van de smelt. De samenstelling van de smelt kan veranderen
tijdens het kristallisatieproces en bijkomende intermetallische verbindingen met
andere samenstellingen kunnen uitkristalliseren.
Welk geval zich voordoet kan uit fasediagrammen worden afgelezen (zie Hoofdstuk 5). Zoals
in Hoofdstuk 4 wordt besproken, hangt de neiging om vaste oplossingen te vormen van een
aantal factoren af, waaronder het chemisch verband tussen de elementen en de relatieve
grootte van hun atomen.
Twee metalen die chemisch met elkaar verwant zijn en waarvan de atomen bijna dezelfde
grootte hebben, vormen wanordelijke legeringen met elkaar. Voor onbeperkte oplosbaarheid
is het ook van belang dat de metalen dezelfde kristalstructuur hebben. Bijvoorbeeld, zilver en
goud hebben beide een kubische dichtste bolstapeling als kristalstructuur en hun atomen zijn
bijna even groot (de straal van een goudatoom bedraagt 144,2 pm en de straal van een
zilveratoom 144,4 pm). Deze metalen vormen met elkaar vaste oplossingen (mengkristallen)
met een arbitraire samenstelling waarin de zilver- en goudatomen willekeurige posities
innemen in het kristalrooster van de kubische dichtste bolstapelingsstructuur. Goud en zilver
zijn in alle verhoudingen mengbaar. Een goud-zilverlegering wordt elektrum genoemd.
Verwante metalen, zeker deze die in dezelfde groep in het periodiek systeem vormen vaste
oplossingen in alle mengverhoudingen als hun atoomstralen niet meer dan 15% van elkaar
verschillen. Voorbeelden zijn Mo + W, Ru + Os, Rh + Pd + Ir + Pt, K + Rb, K + Cs, maar niet
Na + Cs. Als de elementen minder met elkaar verwant zijn, dan kan er een beperkte
91
mengbaarheid voorkomen. Voorbeelden zijn koper en zink: er lost maximaal 34,8 wt.% zink
in koper op, en maximaal 2,3 wt.% koper in zink. Zinkhoudende koperlegeringen noemt ment
messing.
Wanneer de atomen in grootte van elkaar verschillen of wanneer de metalen chemisch
meer verschillend zijn, is de kans groter dat er structuren met een geordende atoomverdeling
voorkomen. Een geordende verdeling van bollen van verschillende groottes laat een betere
ruimtevulling toe dan een verdeling van bollen met éénzelfde grootte. De atomen komen
hierdoor dichter bij elkaar, waardoor de aantrekkingskrachten efficiënter worden. Bijgevolg
zullen metalen met verschillende atoomstralen bij voorkeur in welbepaalde stoechiometrische
verhoudingen in de vaste toestand combineren: ze vormen intermetallische verbindingen.
Zelfs wanneer een volledige mengbaarheid in de vaste toestand mogelijk is, zullen georderde
structuren de voorkeur krijgen als de metaalatomen verschillende groottes hebben.
Bijvoorbeeld koperatomen (straal 127,8 pm) zijn kleiner dan goudatomen (straal 144,2 pm).
Goud en koper vormen in alle verhoudingen mengkristallen, maar er worden bovendien
geordende legeringen gevormd met de samenstellingen AuCu en AuCu3. De mate van orde is
temperatuursafhankelijk: met toenemende temperaturen daalt de mate van ordening continu.
Bij hoge temperaturen worden deze geordende legeringen geleidelijk omgezet tot
wanordelijke legeringen waarbij alle atoomposities statistisch door goud of koper bezet zijn.
3.4 Structuren van covalente kristallen
In moleculaire kristallen houden covalente bindingen de atomen samen ter vorming van
moleculen. De zwakke van de Waalskrachten zorgen voor cohesie tussen de moleculen
onderling ter vorming van zachte kristallen met lage smelt- en kookpunten. In covalente
kristallen binden grotendeels covalente bindingen elk atoom aan de naburige atomen en
bestaan er geen zwakke bindingen. Het gehele kristal kan als een enkelvoudig gigantisch
molecule worden beschouwd. Bijvolg kunnen covalente kristallen zeer hard zijn en hebben ze
hoge smeltpunten. Door de gerichte aard van de covalente binding, komen vooral structuren
voor waarbij de atomen een laag coördinatiegetal hebben en waarbij de atomen los gestapeld
zijn met veel open ruimte tussen de atomen. Daardoor hebben covalente kristallen vaak een
lage massadichtheid.
Diamant is de kubische modificatie van koolstof en is stabiel op hoge temperaturen. In de
diamantstructuur is elk koolstofatoom covalent aan vier omringende koolstofatomen
verbonden, ter vorming van een driedimensionaal netwerk (Figuur 3.9). Elk koolstofatoom is
tetraëdrisch gecoördineerd door zijn vier naburige koolstofatomen. De noodzaak om vier
92
bindingen met andere koolstofatomen aan te gaan, maakt dat koolstof in diamant sp3gehybridiseerd is. Juist zoals in alkanen bedraagt de C-C bindingslengte 154 pm en zijn de
bindingshoeken 109,47°. De eenheidscel van diamant komt overeen met een kubische dichtste
bolstapeling van koolstofatomen (kubisch vlakkengecenterde structuur), met de helft van de
tetraëdrische holten opgevuld. Het aantal koolstofatomen in de eenheidscel van diamant
bedraagt acht. Naast de vier koolstofatomen die zich binnenin de eenheidscel in de
tetraëdrische holten bevatten, dragen de koolstofatomen op de hoekpunten en de vlakken van
de eenheidscel nog een voor vier koolstofatomen per eenheidscel bij. Merk op dat een
koolstofatoom op een hoekpunt van de kubus door 8 andere eenheidscellen worden gedeeld
en dat een koolstofatoom op een vlak tussen 2 eenheidscellen worden verdeeld: 8 hoekpunten
(8× 1/8) en 6 vlakken (6× ½) geeft de hierboven vermelde vier koolstofatomen. Aangezien elk
octant een kubus is met vier van zijn hoekpunten bezet door een koolstofatoom, heeft elke
koolstofatoom in het middelpunt van het octant een tetraëdrische omringing. Dezelfde
redening kan voor de andere koolstofatomen gemaakt worden omdat ze allen symmetrieequivalent zijn. De diamantstructuur wordt ook waargenomen voor silicium, germanium en
grijs tin.
Figuur 3.9. Diamantstructuur.
Sfaleriet (zinkblende) is een kubisch modificatie van zinksulfide. De structuur van
sfaleriet lijkt op deze van diamant, met het verschil dat in sfaleriet elk zinkatoom door vier
zwavelatomen is omringd en vice-versa (Figuur 3.10). De zwavelatomen nemen hierbij de
posities in een kubische dichtste bolstapeling in, waarbij de helft van de tetraëdrische holten
door zinkatomen zijn bezet. De sfalerietstructuur kan ook beschouwd worden als bestaande
uit twee in elkaar gedrongen kubisch vlakkengecenterde kristalroosters, waarbij de
zwavelatomen de roosterplaatsen van het ene kristalrooster innemen en de zinkatomen de
roosterplaatsen in het andere kristalrooster. Andere verbindingen met deze structuur zijn ZnSe
(stilliet), ZnTe, α-CdS (hawleyiet), HgS (metacinnaberiet), HgSe (tiemanniet), HgTe
93
(coloradoïet), CuF, CuCl (nantokiet), CuBr, CuI, GaAs, GaP, InP, BN en SiC. Merk op dat
het hier steeds gaat over binaire verbindingen (2 componenten). Alhoewel de
sfalerietstructuur hier als een covalent kristal wordt beschreven, is een beschrijving als een
ionische verbinding ook mogelijk. Deze reden is dat bij zinksulfide het ionisch of covalent
karakter niet erg uitgesproken zijn.
Figuur 3.10. Sfalerietstructuur. De witte bollen stellen zinkatomen voor en de zwarte bollen
zwavelatomen.
In plaats van in de kubische sfalerietstructuur, kan ZnS ook uitkristalliseren in de
hexagonale structuur van het mineraal wurtziet (Figuur 3.11). In de wurtzietstructuur is elk
zinkatoom door vier zwavelatomen omgeven en vice-versa, zoals in de sfalerietstructuur.
Terwijl de sfalerietstructuur is afgeleid van een kubische dichtste bolstapeling, is de
wurtzietstructuur van een hexagonale dichtste bolstapeling afgeleid. De zwavelatomen
bezetten de posities in een hexagonale dichtste bolstapeling en de zinkatomen bezetten de
helft van de tetraëdrische holten. Wanneer men de sfaleriet- met de wurtzietstructuur
vergelijkt dan merkt men op dat de symmetrie van kationen en anionen ten opzichte van hun
naaste buren gelijk is, maar dat de tweede coördinatiesfeer verschillend is. De
wurtzietstructuur komt voor bij ZnO (zinkiet), BeO (bromelliet), β-CdS (greenockiet), βCdSe (cadmoseliet), GaN, InNn MnS, AgI (iodargyriet), AlN en NH4I. Juist zoals de
sfalerietstructuur, kan de wurtzietstructuur ook als een ionisch rooster worden beschreven. In
Figuur 3.12 worden de verschillen in conformatie van de zwavelatomen rond de zinkatomen
in de sfaleriet- en in de wurtzietstructuur getoond.
94
Figuur 3.11. Wurtzietstructuur. De witte bollen stellen de zinkatomen voor en de zwarte
bollen de zwavelatomen.
Figuur 3.12. Verschillen in conformatie in (a) de sfalerietstructuur en (b)
de wurtzietstructuur.
95
3.5 Structuren van ionische verbindingen
3.5.1 Straalverhoudingen en coördinatiepolyeders
In een eerste benadering bestaan de kristalstructuren van ionische verbindingen uit een
dichtste bolstapeling van relatief grote ionen (gewoonlijk de anionen) waarbij kleine ionen
(gewoonlijk de kationen) een deel van de octaëdrische of tetraëdrische holten tussen de grote
ionen opvullen. In zulk een structuur worden enkel kationen opgenomen die voldoende groot
zijn zodat de kationen niet “rammelen” in de holten tussen de anionen. Deze vereiste dat het
kation contact moet maken met de omringende anionen zorgt ervoor dat de ionen op zulk een
manier gestapeld worden dat de potentiële energie van het kristal wordt geminimaliseerd. Als
een naburig kation en anion elkaar in de structuur niet raken, dan zullen beide een grotere
potentiële energie bezitten dan als ze elkaar wel raken. Vergelijk dit met een magneet en een
ijzeren staaf die elkaar kunnen raken of uit elkaar worden gehouden.
Figuur 3.13. Twee typen van een twaalfvoudige coördinatie van X-ionen rond een A-ion,
waarbij de ionen X en A even groot zijn. In (A) wordt een hexagonale dichtste bolstapeling
getoond en in (D) een kubische dichtste bolstapeling.
Het maximaal aantal grote ionen (X) die een tweede ion (A) kan omringen en raken is
afhankelijk van de relatieve grote van beide ionen, die door de straalverhouding RA/RX wordt
weergegeven (waarbij RA en RX de ionstralen van ion A en X zijn). Als A en X even groot
zijn, d.w.z. als RA/RX = 1, dan kunnnen maximaal 12 X-ionen rond A worden geplaatst zodat
96
de X-ionen elkaar en A raken. De 12 X-ionen kunnen op twee verschillende wijzen rond het
A-ion worden geplaatst, zoals in Figuur 3.11 wordt weergeven. Als het A-ion door een X-ion
zou vervangen worden, dan komt mogelijkheid A in Figuur 3.13 overeen met een hexagonale
dichtste bolstapeling, terwijl B met een kubische dichtste bolstapeling overeenkomt. De
coördinatiepolyeder is verschillend in beide gevallen, een anti-kuboctaëder voor de
hexagonale dichtste bolstapeling en een kuboctaëder voor de kubische dichtste bolstapeling
(zie ook hogerop), maar het coördinatiegetal voor ion A is in beide gevallen 12.
Als het A-ion dat in Figuur 3.13 wordt getoond kleiner zou worden, dan zou het in zijn
site beginnen te “rammelen”. Als de straalverhouding RA/RX kleiner dan 1 wordt, kunnen
slechts 8 X-ionen een A-ion omringen en raken. Het A-ion wordt hierbij omgeven door een
kubus van X-ionen (Figuur 3.14). Als RA/RX nog kleiner wordt, d.w.z. als kleinere A-ionen
tussen de X-ionen worden gestapeld, zullen de X-ionen die zich op de hoekpunten van de
kubus bevinden steeds dichter bij elkaar komen totdat ze elkaar raken. De afstand tussen twee
middelpunten van aangrenzende X-ionen 2RX is dan gelijk aan de lengte a van de zijde van de
kubus. De lengte van de ruimtediagonaal van de kubus is dan 2RX + 2 RA. Aangezien voor een
kubus geldt dat de ruimtediagonaal gelijk is aan √3a, vindt men:
2RX + 2RA = √3(2RX)
Als men beide leden van de vergelijking deelt door 2RX, en de vergelijking oplost naar RA/RX,
verkrijgt men:
RA/RX = √3 – 1 = 0,732
Bijgevolg, als de straalverhouding kleiner wordt dan 0,732, zal het ion A beginnen
“rammelen” in de holte van de omringende X-ionen. In dat geval is de achtvoudige
coördinatie van A-ionen door X-ionen niet meer de plaatsing van ionen met de laagste
potentiële energie.
97
Figuur 3.14. Een achtvoudige coördinatie van acht X-ionen rond een A-ion wordt begunstigd
voor straalverhoudingen RA/RX tussen 0,732 en 1.
Voor een straalverhouding tussen 0,732 en 0,414 kunnen slechts zes X-ionen een A-ion
omringen (Figuur 3.15). Deze zes X-ionen vormen een octaëder rond A en daarom spreekt
men van een octaëdrische coördinatie. De onderlimiet, waaronder het kation in de
anionenholte begint te “rammelen” (straalverhouding 0,414), is voor een octaëdrische
omgeving is dezelfde als wanneer vier grote X-ionen op de hoeken van een vierkant rond het
ion A worden geplaatst. De zijde van het vierkant dat wordt bepaald door de middelpunten
van de X-ionen heeft een lengte die gelijk is aan 2RX. Bijgevolg is de lengte van de diagonaal
gelijk aan √2RX. De diagonaal is echter ook gelijk aan 2RX + 2 RA, zodat:
2RX + 2RA = √2(2RX)
Als men beide leden van de vergelijking deelt door 2RX, en de vergelijking oplost naar RA/RX,
verkrijgt men:
RA/RX = √2 – 1 = 0,414
Figuur 3.15. Octaëdrische omringing van zes X-ionen rond een ion A wordt begunstigd als de
straalverhouding RA/RX tussen 0,732 en 0,414 gelegen is.
98
Een tetraëdrische omgeving van vier X-ionen rond een A-ion heeft men voor
straalverhoudingen RA/RX tussen 0,414 en 0,225 (Figuur 3.16). Voor straalverhoudingen
tussen 0,225 en 0,155 heeft men een driehoekige coördinatie van drie X-ionen in één vlak
rond een A-ion.Voor straalverhoudingen kleiner dan 0,155 is het grootteverschil tussen X en
A zo groot dat er maximaal twee X-ionen rond een A-ion kunnen geplaatst worden zodat er
aan de voorwaarde is voldaan dat alle omringende X-ionen het ion A raken.
Figuur 3.16. (A) tetraëdrische coördinatie van vier X-ionen rond een A-ion voor
straalverhoudingen RA/RX tussen 0,414 en 0,225; (B) drievoudige coördinatie van drie Xionen rond een A-ion voor straalverhoudingen RA/RX tussen 0,225 en 0,155;
(C) lineaire coördinatie van twee X-ionen rond een A-ion voor straalverhoudingen RA/RX
kleiner dan 0,155.
Alle hierboven beschouwde coördinatiepolyeders (kubus, octaëder, tetraëder,...) zijn
regelmatige
coördinatiepolyeders.
Men
kent
echter
ook
veel
onregelmatige
coördinatiepolyeders, waarbij niet alle grote X-ionen zich op eenzelfde afstand van het
kleinere centrale kation A bevinden. Sommige van deze onregelmatige coördinatiepolyeders
zijn slechts kleinere verstoringen van de regelmatige polyeders en het coördinatiegetal van het
kation A is hierbij 2,3, 4, 6, 8 of 12. Andere coördinatiepolyeders zijn onregelmatige
polyeders waarbij 5, 7, 9 of 10 anionen op verschillende afstanden rond het centrale kation
worden geplaatst. De anionen (of atomen) die raken aan het central kation, noemt men de
naaste buren en samen vormen ze de eerste coördinatieschil. De ionen (of atomen) die aan
deze naaste buren raken, worden de volgende naaste buren genoemd en samen vormen ze de
tweede coördinatieschil. De bindingskrachten tussen het centrale metaalion en de ionen (of
atomen) in de eerste coördinatieschil zijn relatief sterk. De krachten tussen het centrale
metaalion en de ionen (of atomen) in de volgende coördinatieschillen worden snel veel
kleiner.
99
3.5.2 Structuren van binaire ionische verbindingen
Kristallen waarbij de ionen A en X een gelijke, maar tegengestelde elektrische lading
hebben, bevatten een gelijk aantal ionen A en X om elektrische neutraliteit te verkrijgen en
kunnen door de algemene formule AX worden voorgesteld. Als de straalverhouding RA/RX
groter dan 0,732 is, wordt een coördinatie van acht X-ionen rond een A-ion begunstigd. Dit
wordt verkregen als de X-ionen een primitief kubisch rooster vormen en de A-ionen de
relatief grote kubische holten tussen de X-ionen vullen. Dit wordt in de CsCl-structuur
waargenomen (Figuur 3.16). Elk X-ion wordt ook door acht A-ionen omgeven. De CsClstructuur vindt men voor CsCl (0,92) CsBr (0,86), CsI (0,76) en NH4Cl (0,79) (tussen haakjes
staat de straalverhouding RA/RX weergegeven).
Figuur 3.16. (A) Vier eenheidscellen van de CsCl-structuur; (B) Coördinatie van acht Aionen rond een X-ion in de CsCl-structuur.
Als de straalverhouding RA/RX tussen 0,732 en 0,414 gelegen is, heeft men een
octaëdrische coördinatie van zes X-ionen rond een A-ion. Ionische AX-structuren verkrijgen
deze coördinatie door een kubische dichtste bolstapeling van X-ionen, waarbij de kleinere Aionen de octaëdrische holten bezetten. Dit is in de NaCl-structuur waar te nemen (Figuur
3.17). In de NaCl-structuur is elk A-ion octaëdrisch omgeven door zes X-ionen en is elk Xion octaëdrisch omgeven door zes A-ionen. De grotere chloride-ionen zijn volgens een
kubische dichtsted bolstapeling gestapeld en de kleinere natriumionen bezetten de
octaëdrische holten. Alhoewel Cl- en Na+ eenzelfde aantal elektronen hebben, zijn de Na+
ionen kleiner omwille van de positieve lading. Meer dan 200 verbindingen kristalliseren in de
NaCl-structuur uit, waaronder: LiF (0,57), LiCl (0,42), LiBr (0,39), LiI (O,35), NaF (0,73),
NaCl (0,54), NaBr (0,50), NaI (0,44), KF (1,00), KCl (0,73), KBr (0,68), KI (0,60), RbF
(1,11), RbCl (0,81), RbBr (0,75), RbI (0,67), CsF (1,26), AgF (0,95), AgCl (0,70), AgBr
100
(0,65), MgO (0,47), CaO (0,71), SrO (0,80), BaO (0,96), FeO (0,53), CoO (0,51), NiO (0,49),
MgS (0,36), CaS (0,54), SrS (0,61), BaS (0,73), MnS (0,43) en PbS (0,65) (tussen haakjes
staat de straalverhouding RA/RX weergegeven). Men merkt op dat voor verscheidene
verbindingen de straalverhouding buiten de limiet 0,732 > RA/RX > 0,414 valt. Dit wijst er op
dat er naast de straalverhouding nog andere factoren een invloed hebben op het type van
kristalstructuur dat door een verbinding wordt aangenomen. In een aantal verbindingen heeft
de chemische binding een grote covalente of metallische bijdrage, en in deze gevallen zijn de
straalverhoudingen minder van belang. Bij het bepalen van de straalverhoudingen neemt men
aan dat de ionen harde niet-vervormbare bollen zijn. Dit is natuurlijk niet altijd het geval.
Vele zachte Lewiszuren en Lewisbasen hebben sterk vervormbare ionenwolken.
Figuur 3.17. NaCl-structuur.
AX-verbindingen waarbij de chemische binding voor een aanzienlijk gedeelte een
metaalbinding heeft, hebben vaak de NiAs-structuur (Figuur 3.18). Hierbij nemen de
arseenatomen een hexagonale dichtste bolstapeling aan en de nikkelatomen bezetten de
octaëdrische holten tussen de arseenatomen. Elk nikkelatoom wordt octaëdrisch door zes
arseentatomen omgeven, terwijl elk arseenatoom omgeven wordt door zes nikkelatomen die
zich op de hoekpunten van een trigonaal prisma bevinden. De nikkelatomen in twee
opeenvolgende lagen in de kristalstructuur liggen exact boven elkaar. Dit is ook een
aanduiding dat ze de (octaëdrische) holten vullen en dat de nikkelatomen niet volgens een
dichtste bolstapeling gestapeld zijn. Bij dichtste bolstapelingen komt een AAA stapeling met
alle atomen vlak boven elkaar immers niet voor. Binnenin de nikkellaag hebben de
nikkelatomen wel de omringing die ook bij een dichtste bolstapeling voorkomt. De NiAsstructuur wordt waargenomen voor de mineralen niccoliet (NiAs), breithauptiet (NiS),
101
pyrrhotiet (FeS), evenals voor CoS, VS en voor verbindingen waarbij een transitiemetaal
wordt gecombineerd met Sn, Pb, As, Sb, Bi, Se of Te zoals FeSe, FeTe, FeSb, CrSe, CrTe,
CoSe, MnSb, NiSb, PtSn en PtBi. Zuiver ionische verbindingen met een straalverhouding
RA/RX tussen 0,732 en 0,414 nemen gewoonlijk de NaCl-structuur aan en niet de NiAsstructuur.
Figuur 3.18. De NiAs-structuur. De witte bollen stellen de arseenatomen voor en de zwarte
bollen de nikkelatomen.
Voor een straalverhouding RA/RX tussen 0,414 en 0,225 nemen de AX-verbindingen de
sfaleriet- of de wurtzietstructuur aan. Deze structuurtypen werden reeds hogerop beschreven
als afgeleiden van de diamantstructuur. Zoals reeds vermeld kunnen deze structuren ook als
ionische structuurtypen worden beschreven, omdat de chemische binding in ZnS grotendeels
covalent is. In beide structuren omgeven vier X-ionen tetraëdrisch een A-ion. Als de binding
grotendeels covalent is, kunnen afwijkingen van de limieten opgelegd door de
straalverhoudingen voorkomen. Bijvoorbeeld, ZnO (zinkiet) kristalliseert uit in de
wurtzietstructuur, alhoewel de straalverhouding 0,53 bedraagt en men voor deze verbinding
de NaCl-structuur verwacht. Door de covalentie van de bindingen wordt de wurtzietstructuur
aangenomen.
Er kunnen 12 anionen rond een kation worden gerangschikt als de straalverhouding tussen
0,95 en 1,00 is. Deze coördinatie komt echter niet bij ionische verbindingen voor, omdat in dit
geval het niet mogelijk is om tegelijkertijd kationen enkel door anionen te omringen en
anionen door kationen. Als de verhouding RA/RX groter wordt dan 1, zoals dit het geval is
voor RbF en CsF, worden de relaties omgekeerd: de kationen zijn groter dan de anionen en de
contacten tussen de kationen bepalen nu de beperkende straalverhouding. Dezelfde numerieke
waarden en structuurtypes zijn van toepassing, maar nu moet de omgekeerde straalverhouding
RX/RA worden beschouwd.
102
Voor de structuurtypes voor de AX-verbindingen die we tot hiertoe hebben vermeld,
hebben anionen en kationen eenzelfde coördinatiegetal. In het geval van AX2-verbindingen is
het coördinatiegetal van de kationen tweemaal zo groot als dat van de anionen. Zulk een X/Averhouding kan verkregen worden als de X-ionen zich volgens een dichtste bolstapeling
stapelen en de A-kationen slechts 50% van de octaëdrische holten of 25% van de tetraëdrische
holten
bezetten.
De
geometrische
beschouwingen
betreffende
de
relaties
tussen
straalverhoudingen en coördinatiepolyeders blijven dezelfde. Twee structuurtypes vervullen
de vereiste voorwaarden en komen veel voor bij AX2-verbindingen: het fluoriettype (CaF2) en
het rutieltype (TiO2).
AX2-verbindingen kristalliseren in de fluorietstructuur uit als de straalverhouding RA/RX
groter dan 0,732 is. In de fluorietstructuur is elk kation A door 8 anionen X omgeven in een
kubische coördinatie, terwijl elk anion X tetraëdrisch door 4 kationen A omgeven is (Figuur
3.19). De fluorietstructuur lijkt op de CsCl-structuur, behalve dat slechts om de andere kubus
die gevormd wordt door de primitief kubische stapeling van X-ionen een A-ion in zijn
middelpunt heeft. In deze structuur, die in feite deze van het mineraal fluoriet (CaF2) is, deelt
een kubus met een A-ion in zijn middelpunt enkel zijn vlakken met lege kubussen. Bijgevolg
worden kation-kation-repulsies tussen de tweewaardige A2+-kationen geminimaliseerd. De
eenheidscel van fluoriet heeft een kubische symmetrie. De fluorietstructuur kan worden
beschouwd als een kubische dichtste bolstapeling van de calcium-ionen, waarbij de fluorideionen alle tetraëdrische holten bezetten. Omdat er tweemaal zoveel tetraëdrische holten dan
bollen zijn in een kubische dichtste bolstapeling, is de stoechiometrie CaF2. Dit wordt in
Figuur 3.19b duidelijk getoond. Merk op dat de Ca2+ ionen (ionstraal: 99 pm) groter zijn dan
de F- ionen (ionstraal: 71 pm). Voorbeelden van andere verbindingen die in de
fluorietstructuur uitkristalliseren zijn SrF2, BaF2, EuF2, CdF2, PbF2, SrCl2, BaCl2, ZrO2, HfO2,
CeO2 (cerianiet), ThO2 (thorianiet) en UO2 (uraniniet).
103
Figuur 3.19. Fluorietstructuur. Merk op dat in tegenstelling tot wat in deze figuur wordt
getoond de calciumionen (ionstraal: 99 pm) groter zijn dan de fluoride-ionen
(ionstraal: 71 pm).
Voor AX2-verbindingen heeft men de rutielstructuur als de straalverhouding RA/RX tussen
0,414 en 0,732 gelegen is. In de rutielstructuur, die voorkomt bij het mineraal rutiel (TiO2), is
elk A-kation octaëdrisch omgeven door 6 X-anionen, en is elk anion omgeven door een
driehoek van A-kationen (Figuur 3.20). De rutielstructuur kan beschreven worden als een
hexagonale dichtste bolstapeling van X-ionen, waarbij de A-kationen de helft van de
octaëdrische sites bezetten. De eenheidscel van rutiel heeft een tetragonale symmetrie. De
structuur geeft de sterke neiging van het Ti4+-ion om een octaëdrische omringing aan te
nemen weer. Verbindingen die ook in de rutielstructuur uitkristalliseren zijn: GeO2, MnO2,
RuO2, CoO2, PbO2 (plattneriet), SnO2 (cassiteriet), TeO2, MgF2, FeF2, ZnF2 en MnF2.
104
Figuur 3.20. Rutielstructuur.
Als men de posities van anionen en kationen omwisselt bij de CsCl-, NaCl-, of ZnSstructuren, dan heeft de resulterende structuur dezelfde structuur als de oorspronkelijke
structuur. Als men in het geval van een verbinding met een fluorietstructuur anionen voor
kationen uitwisselt en vice-versa, krijgt men ook een uitwisseling van de coördinatiegetallen,
d.w.z. dat de anionen nu een coördinatiegetal 8 krijgen en de kationen een coördinatiegetal 4.
Dit structuurtype noemt men soms een “anti-fluorietstructuur” (Figuur 3.21). Deze komt voor
bij de alkalimetaaloxiden Li2O, Na2O, K2O, Rb2O en Cs2O, evenals bij de sulfiden, seleniden
en telluriden van de alkalimetalen.
Figuur 3.21. Anti-fluorietstructuur.
De CdI2-structuur is een voorbeeld van een structuurtype met een uitgesproken
laagstructuur (Figuur 3.22). De jodide-ionen vormen een bijna perfecte hexagonale dichtste
bolstapeling. De anionlagen worden samengehouden door de Cd2+-kationen die de helft van
de octaëdrische holten bezetten. De bezetting van de octaëders is niet willekeurig, aangezien
105
er in de kristalstructuur afwisselend lagen voorkomen met volledig bezette octaëderholten en
lagen met volledig onbezette octaëderholten. Hierdoor is de structuur opgebouwd uit “XAX”sandwichen, bestaande uit een laag met A-ionen met een laag aan X-ionen aan beide zijden.
Terwijl de binding tussen de A- en X-ionen ionisch is, worden de XAX-sandwichen onderling
slechts door zwakke van der Waalskrachten bij elkaar gehouden. Het gevolg is dat
verbindingen met de CdI2-structuur gemakkelijk splijten volgens de richting van de XAXlagen. Dit structuurtype komt zeer veel voor bij de bromiden, jodiden en hydroxiden van
tweewaardige metalen, evenals bij de sulfiden, seleniden en telluriden van vierwaardige
metalen. Voorbeelden zijn: CaI2, Ca(OH)2 (portlandiet), MgBr2, MgI2, Mg(OH)2 (bruciet),
FeBr2, FeI2, CoBr2, CoI2, TiS2, ZrS2, SnS2, SnSe2, PtS2, PtSe2, PtTe2, IrTe2. Bij de verwante
CdCl2-structuur nemen de anionen een kubische dichtste bolstapeling aan, en bezetten de
kationen de helft van de octaëderholten. Dit structuurtype komt voor bij MgCl2
(chloromagnesiet), FeCl2, MnCl2, CdCl2, CdBr2, CoCl2, NiCl2, NiBr2 en NiI2.
Figuur 3.22. CdI2-structuur.
3.5.3 Structuur van ternaire ionische verbindingen
Wanneer er drie verschillende bolvormige ionen aanwezig zijn, dan zijn hun relatieve
groottes ook een belangrijke factor die de stabiliteit van de structuur controleert. De
tetragonale PbFCl-structuur (matlockiet) is een voorbeeld van een structuur waarbij de
anionen naargelang hun groottes een verschillende bezettingsgraad in de atoomlagen vertonen
106
(Figuur 3.23). De chloride-ionen vormen een laag met een vierkant patroon. Bovenop de
chloridelaag bevindt zich een laag fluoride-ionen, ook in een vierkant patroon, maar over 45°
gedraaid. De fluoride-ionen zijn gesitueerd boven de zijden van de vierkanten die door de
chloride-ionen worden gevormd. Door deze stapeling zijn de F-F afstanden een factor 0,707
(= ½√2) korter dan de Cl-Cl afstanden. Dit komt overeen met de straalverhouding RF-/RCl- =
0,73. Een fluoridelaag bevat dubbel zoveel ionen als een chloridelaag. Elke vier fluorideionen en elke vier chloride-ionen vormen een antiprisma, waarin zich een Pb2+-ion in het
centrum bevindt. De Pb-F afstanden zijn duidelijk korter dan de Pb-Cl afstanden, waardoor
het vierkant gevormd door de vier fluoride-ionen kleiner is dan het vierkant gevormd door de
vier chloride-ionen. Onder de helft van de vierkanten in de fluoridelaag bevindt zich een Pb2+ion, terwijl bij de andere helft van de vierkanten in de fluoridelaag het Pb2+-ion zich boven de
laag bevindt. De antiprisma’s worden vervolledigd door de chloride-ionen die zich bevinden
in de chloridelaag die zich boven en onder de fluoridelaag zich bevindt. Het aantal Pb2+-ionen
is gelijk aan het aantal fluoride-ionen. Het aantal chloride-ionen is even groot als het aantal F-ionen, omdat er voor elke fluoridelaag er twee chloridelagen aanwezig zijn. In de
kristalstructuur komen laagpakketten voor bestaande uit een fluoridelaag omgeven door twee
chloridelagen. De lagen worden zodanig gestapeld dat de chloride-ionen zich plaatsen in de
holten van de onderliggende chloridelaag. Hierdoor wordt de coördinatiesfeer van het Pb2+ion met een bijkomend chloride-ion uitgebreid zodanig dat het coördinatiegetal van een Pb2+ion negen is. Vele verbindingen hebben de PbFCl-structuur. De omvatten de grote klasse van
de oxyhaliden MOX (M = Bi, lanthanide, actinide, X = Cl, Br, I), hydridehaliden zoals CaHCl
en vele metallische verbindingen zoals NbSiAs.
Figuur 3.23. PbFCl-structuur
107
Figuur 3.24. Perovskietstructuur.
Vele verbindingen met een ABX3-stoechiometrie nemen de structuur van perovskiet
(CaTiO3) aan. De perovskietstructuur bestaat in eerste benadering uit een kubische dichtste
bolstapeling van oxide-ionen, waarbij calcium-ionen de plaats innemen van éénvierde van de
oxide-ionen (Figuur 3.24). De titaan(IV)-ionen bezetten éénvierde van de octaëdrische holten
in deze kubische dichtste bolstapeling van oxide- en calciumionen, waarbij de octaëdrische
holten die niet naast een calcium-ion liggen worden bezet. De conventionele keuze van de
eenheidscel voor perovskiet is diegene waarbij de oorsprong van het kristalrooster gelegen is
op de titaan(IV)ionen. De Ti-atomen bevinden zich dan op de hoekpunten van de eenheidscel,
het Ca-atoom in het middelpunt en de O-atomen in het midden van de ribben van de kubus.
Deze keuze toont echter minder duidelijk dat de oxide- en calciumionen bij benadering een
kubische dichtste bolstapeling vertonen, d.w.z. dat ze een kubisch vlakkengecenterd rooster
vormen. De calciumatomen zijn omgeven door 12 zuurstofatomen, terwijl de titaanatomen
door 6 zuurstofatomen omgeven zijn. Elk zuurstofatoom raakt vier calciumatomen en twee
titaanatomen. Als Ca2+ even groot zou zijn als O2-, en dus perfect in een twaalfvoudig
gecoördineerde site zou passen, dan zou de afstand tussen overstaande twee O2--ionen in het
vierkant met het Ca2+-ion als middelpunt gelijk zijn aan 2RO + 2RCa, of 1,414× de lengte van
de ribbe van de kubische éénheidscel. De ribbe van de éénheidscel is ook gelijk aan 2RO +
2RTi. Bijgevolg is:
RO + RCa = 1,414(RO + RTi)
Dit is de ideale verhouding tussen de ionstralen van de ionen in een perovskietstructuur. De
structuur verdraagt echter een zekere variatie, zodat deze uitdrukking kan aangepast worden
tot:
RO + RCa = t⋅1,414(RO + RTi)
108
Hierbij is t de zogenaamde tolerantiefactor en de waarde van t is gewoonlijk tussen 0,8 en 1,0
gelegen. Als X O2--ionen in deze ABX3-structuur voorstelt, moeten de valenties van A en B in
totaal gelijk zijn aan zes. Mogelijke combinaties zijn dus 1 + 5, 2 + 4 of 3 + 3. Als X een F-ion voorstelt, moeten de valenties van A en B in totaal gelijk zijn aan drie of 1 +2. De ideale
kubische perowskiet-structuur die in Figuur 3.21 wordt getoond komt enkel voor bij de
weinige verbindingen waarbij de tolerantiefactor tussen 0,90 en 1,0 gelegen is. Voorbeelden
zijn KNbO3 (0,93), BaSnO3 (0,92), KMgF3 (0,95), KZnF3 (0,91), PbZnF3 (0,90) en KNiF3
(0,93) (tussen haakjes wordt de tolerantiefactor gegeven). Voor tolerantiefactoren kleiner dan
0,90 worden verstoorde perovskietstructuren gevormd, waarbij de atomen een beetje
verplaatst zijn van de ideale posities, zodat de symmetrie van de kristalstructuur wordt
verlaagd. Perovskiet zelf (t = 0,81) komt onder een orthorhombische of monokliene vorm
voor en is dus pseudo-kubisch. De afwijking is echter klein: één van de hoeken van de
eenheidscel is 90°40’ in plaats van 90°. De meest voorkomende distorsie is een tilt van de
BO6-octaëders.
Figuur 3.25. Spinelstructuur.
De spinelstructuur bestaat in essentie een kubische dichtste bolstapeling van oxide-ionen
waarbij de metaalionen 1/8 van de tetraëdrische holten en 1/2 van de octaëdrische holten
bezetten (Figuur 3.25). Bij de normale spinellen bezetten tweewaardige metaalionen A2+ de
tetraëdrische holten en driewaardige ionen B3+ bezetten de octaëdrische holten. De algemene
formule is dan AB2O4 en de eenheidscel bevat 32 zuurstofatomen 8 A-atomen en 16 Batomen. Bij de inverse spinellen verwisselen de A2+-ionen van plaats met de helft van de B3+-
109
ionen. De algemene formule is bijgevolg B[AB]O4. Zowel spinellen als inverse spinellen
hebben een kubische symmetrie. Voorbeelden van verbindingen met de spinelstructuur zijn:
MgAl2O4 (spinel), Fe2+Al2O4 (hercyniet), ZnAl2O4 (gahniet), MnAl2O4 (galaxiet), Fe2+Cr2O4
(chromiet), NiFe23+O4 (trevoriet), MnFe23+O4 (jacobsiet), ... Voorbeelden van verbindingen
met een inverse spinelstructuur zijn: Fe3+[MgFe3+]O4 (magnesioferriet) en Fe3+[Fe2+Fe3+]O4
(magnetiet). Magnetiet, Fe3O4, is een interessante verbinding omdat het een element in twee
oxidatietoestanden bevat.
3.6 Structuren van verbindingen met complexe ionen
De structuur van ionische verbindingen met complexe ionen kan in veel gevallen worden
afgeleid van de structuur van eenvoudige ionische verbindingen. Een bolvormig ion wordt
hierbij door een complex ion vervangen, zodat het kristalrooster op een geschikte wijze moet
vervormd worden om zich aan te passen aan de vorm van het complexe ion.
Staafvormige ionen zoals CN-, C22- of N3- kunnen de Cl--ionen in de NaCl-structuur
vervangen wanneer alle staafvormige ionen parallel aan elkaar worden geörienteerd en
wanneer het kristalrooster in de overeenkomstige richting wordt uitgerokken. In de structuur
van calciumcarbide, CaC2, is het acetylide-ion parallel met één van de zijden van de
eenheidscel. Het gevolg is dat de symmetrie niet langer meer kubisch is, maar wel tetragonaal
(Figuur 3.26). De hyperoxiden KO2, RbO2 en CsO2 evenals peroxiden zoals BaO2
kristalliseren in de calciumcarbide-structuur uit. In de structuren van CsCN en NaN3 zijn het
cyanide- en azide-ion parallel aan één van de ruimtediagonalen van de eenheidscel waardoor
de symmetrie rhomboëdrisch is (Figuur 3.27).
Figuur 3.26. Kristalstructuur van calciumcarbide.
110
Figuur 3.27. Kristalstructuur van natriumazide
De structuur van calciet (CaCO3) kan afgeleid worden van de NaCl-structuur door de Cl-ionen door CO32--ionen te vervangen en Na+ door Ca2+. De carbonaatgroepen staan loodrecht
op één van de ruimtediagonalen van de eenheidscel en vereisen een expansie van het rooster
loodrecht op deze diagonaal (Figuur 3.28). Elk calciumatoom is omgeven door zes
zuurstofatomen. Elke zuurstofatoom is omgeven door twee calciumatomen evenals door het
koolstofatoom van de carbonaatgroep. De symmetrie van calciet is trigonaal. De
kristalstructuur van calciet werd reeds in 1914 door W.L. Bragg bepaald. De calcietstructuur
wordt waargenomen voor andere carbonaten zoals MgCO3 (magnesiet), ZnCO3 (smithsoniet),
CoCO3 (sferocobaltiet), FeCO3 (sideriet), MnCO3 (rhodochrosiet), CdCO3 (otaviet), evenals
voor nitraten zoals LiNO3, NaNO3 (nitronatriet) en voor boraten zoals ScBO3, AlBO3, InBO3,
YBO3. CaCO3 kan ook in de orthorhombische aragonietstructuur uitkristalliseren. Deze
structuur laat zich van de NiAs structuur afleiden, als de As-atomen door Ca-atomen worden
vervangen en de Ni-atomen door CO3-groepen. Terwijl in calciet alle carbonaatgroepen in één
vlak liggen en naar dezelfde richting wijzen, liggen de carbonaatgroepen in aragoniet in twee
verschillende structurele vlakken en wijzen de driehoekige carbonaatgroepen van het ene vlak
in de tegenovergestelde richting van deze van het andere vlak. Elk calciumatoom is door
negen zuurstofatomen omgeven. De stapeling van de kationen benadert deze van een
hexagonale dichtste bolstapeling. De aragonietstructuur komt voor bij SrCO3 (strontianiet),
BaCO3 (witheriet), PbCO3 (cerussiet), KNO3 (nitrokaliet), LaBO3 en NdBO3. De regel is dat
verbindingen waarbij de straal van het kation kleiner is dan deze van Ca2+, met de
calcietstructuur kristalliseren, terwijl verbindingen waarbij de straal van het kation groter is
dan deze van Ca2+, met de aragonietstructuur kristalliseren. CaCO3 heeft de neiging om zowel
in de calciet- als in de aragonietstructuur uit te kristalliseren naar gelang de omstandigheden.
Bij kamertemperatuur en atmosfeerdruk is calciet de stabiele modificatie.
111
Figuur 3.28. Kristalstructuur van calciet.
De structuur van K2PtCl6 kan men van de kristalstructuur van fluoriet afleiden: de
octaëdrische [PtCl6]2--groepen nemen de positie van Ca in en de 8 K+-ionen nemen de posities
van de F--ionen in. In de structuur (Figuur 3.29). In deze structuur maakt elke van een groep
van vier [PtCl6]2--groepen met één octaëderzijde contact met een kaliumion, waardoor het
coördinatiegetal van kalium 12 is.
Figuur 3.29. Kristalstructuur van K2PtCl6.
112
3.7 Silicaatstructuren
In de Natuur zijn silicaten zeer belangrijke verbindingen, aangezien meer dan 80% van de
aardkorst uit silicaatmineralen is opgebouwd. Onafgezien van de verschillende Si/Overhoudingen die in de chemische brutoformules van silicaten tot uitdruk komen (SiO3, SiO4,
Si2O5, Si2O7, Si3O8, Si4O11), bestaat de bouwsteen van silicaten steeds uit de [SiO4]4--tetraëder
(figuur 3.30). Het Si4+-ion bevindt zich in het middelpunt van de tetraëder en is omgeven door
vier O2--ionen. De tetraëder is bijna onverstoord. De Si-O-afstanden zijn tussen de 1,60 Å en
1,64 Å, terwijl de O-O-afstanden (zijden van de tetraëder) variëren tussen 2,50 en 2,70 Å.
Wegens hun grootte (1,40 Å) raken de vier O2--ionen elkaar in de tetraëder, zodanig dat er een
kleine holte voor het Si4+-ion (straal 0,39 Å) ter beschikking staat. De straalverhouding RSi/RO
= 0,278 zodat ook op basis van de regels voor de straalverhoudingen voor ionische structuren
kan voorspeld worden dat silicium door vier zuurstofatomen wordt gecoördineerd. Op basis
van het verschil in elektronegativiteit tussen silicium en zuurstof, kan men afleiden dat de SiO-binding ongeveer 60% covalent en 40% ionisch karakter heeft. Alhoewel de chemisch
binding gedeeltelijk wordt gevormd door de aantrekking tussen de tegengesteld geladen
ionen, is er toch een duidelijke directionele binding in de silicaattetraëder wegens de
ruimtelijke gerichtheid van de sp3-hybride-orbitalen van silicium. De [SiO4]2--tetraëders
hebben in oplossing, in smelten en in kristalstructuren een grote stabiliteit. Men kan
silicaatstructuren beschouwen als dichtste bolstapelingen van oxide-ionen met de Si4+-ionen
in de tetraëdrische holten en met de andere metaalionen in tetraëdrische of in octaëdrische
holten. De silicaten kunnen als verbindingen van metaaloxiden met SiO2 worden opgevat. Zo
ontstaan uit smelten van 2 MgO + SiO2 = Mg2SiO4 olivijn, uit 3CaO + Al2O3 + 3SiO2 =
Ca3Al2[SiO4]3 granaat, uit 2MgO + 2SiO2 = Mg2[Si2O6] enstatiet, uit CaO + Al2O3 + 2SiO2 =
Ca[Al2Si2O6] anorthiet. De zuurstofatomen van het silicaatcomplex kunnen tot twee
verschillende [SiO4]2--tetraëders (maar niet meer) behoren. Hierdoor ontstaan er naast de
geïsoleerde [SiO4]2--tetraëders ook andere bouwmotieven (Figuren 3.31-3.33). Merk op dat de
tetraëders enkel hoekpunten met elkaar kunnen delen, maar nooit zijden of vlakken.
Figuur 3.30. [SiO4]4- tetraëder. A. Ruimtevullend model; B. Polyederrepresentatie.
113
Silicaten met een Si:O-verhouding van 1:4, bestaan uit onafhankelijke [SiO4]4--tetraëders,
naast andere metaalionen. Typische voorbeelden zijn Be2[SiO4] (fenakiet), Zr[SiO4] (zirkoon)
en Mg2[SiO4] (forsteriet). Deze silicaten noemt men nesosilicaten. (Grieks “nesos” = eiland).
De silicaattetraëders zijn met behulp van de kationen van andere elementen tot een
kristalrooster verbonden. Deze metaalionen bevinden zich in de octaëdrische holten van de
structuur. Nesosilicaten vertonen een dichtste stapeling van zuurstofatomen, waardoor ze een
hoge massadichtheid (> 3 g/cm3), een hoge brekingsindex en een grote hardheid hebben.
Silicaten met een Si:O-verhouding van 1:5, hebben naast de SiO4-tetraëders ook nog
tetraëdervreemde O-ionen. Voorbeelden zijn Al2O3 + SiO2 = Al2[O⏐SiO4] (andalousiet) of
CaO + TiO2 + SiO2 = CaTi[O⏐SiO4] (titaniet).
Bij silicaten met een Si:O-verhouding van 1:3,5 kan een tetraëdrische SiO4-coördinatie
enkel worden verkregen als een deel van de O-atomen gelijktijdig met twee Si-atomen
verbonden zijn tot een dubbeltetraëder. [Si2O7]6-. Silicaten met dubbeltetraëders noemt
sorosilicaten (Grieks “soros” = groep). Voorbeelden zijn Sc2[Si2O7] (thortveitiet) Ca3[Si2O7]
(rankiniet) en Ca2Mg[Si2O7] (åkermaniet). De stapeling van de zuurstofatomen is minder
compact dan bij de nesosilicaten.
Als meer dan twee tetraëders worden verbonden, kunnen gesloten ringvormige structuren
worden gevormd met de algemene samenstelling [SixO3x]2x- zoals [Si3O9]6-, [Si4O12]8-,
[Si6O18]12-. Deze silicaten met ringvormige structuren zijn de cyclosilicaten (Grieks “cyclos” =
ring). De structurele eenheid [Si3O9]6- duidt op een driering, zoals in BaTi[Si3O9] (benitoïet),
de eenheid [Si4O12]8- duidt op een vierring zoals in (Ca,Fe,Mn)3Al2(BO3)(Si4O12)(OH)
(axiniet), en [Si6O18]12- wijst op een zesring zoals in Al2Be3[Si6O18] (beryl).
Figuur 3.31. Structuur van de nesosilicaten (enkelvoudige tetraëders), sorosilicaten
(dubbeltetraëders) en cyclosilicaten (ringstructuren).
De silicaattetraëders kunnen ook verbonden worden tot oneindig lange enkelvoudige
ketens met de Si:O-verhouding 1:3 en structurele eenheid [Si2O6]4-. Elke tetraëder deelt twee
114
zuurstofatomen met naburige tetraëders. Voorbeeld is pyroxeen, CaMg[Si2O6]. Bij oneindige
dubbele ketens is de Si:O-verhouding 4:11, en de structurele eenheid [Si4O11]6-. De
tetraëdrisch delen hierbij afwisselend twee en drie zuurstofatomen met naburige tetraëders.
Voorbeeld is antophylliet, (Mg,Fe)7Si8O22(OH)2. Beide vormen van ketenvormige silicaten
zijn bekend onder de benaming inosilicaten (Grieks “inos” = vezel). Wanneer drie zuurstoffen
van een tetraëder met andere tetraëders worden gedeeld, worden oneindig uitgestrekte lagen
gevormd waarbij de Si:O-verhouding 1:2,5 en de structurele eenheid [Si4O10]4-. Deze silicaten
met een bladvormige structuur zijn de phyllosilicaten (Grieks “phyllos” = blad). Voorbeelden
zijn Mg3Si2O5(OH)4 (chrysotiel) en Mg3Si4O10(OH)2 (talk).
Wanneer alle vier zuurstofatomen van de silicaattetraëder met andere tetraëders worden
gedeeld en dus elk zuurstofatoom met twee siliciumatomen wordt gedeeld, ontstaat een
driedimensioneel netwerk met Si:O-verhouding 1:2. Deze silicaten met een netwerkstructuur
zijn de tektosilicaten (Grieks “tektos” = netwerk). Voorbeelden hiervan zijn kwarts, tridymiet
en cristobaliet, die alle drie modificaties van SiO2 zijn. Andere silicaten kunnen enkel een
driedimensioneel netwerk vormen als een deel van de Si4+-ionen door Al3+-ionen vervangen
wordt.
Figuur 3.32. Verschillende ketenstructuren bij de inosilicaten.
115
Figuur 3.33. Structuur van phyllosilicaten (bladstructuur) en tektosilicaten
(netwerkstructuur).
Aangezien de ionstraal van het driewaardige Al3+-ion (0,51 Å ) slechts beperkt groter is
dan die van het Si4+-ion (0,39 Å), kan het Al3+-ion een dubbelrol vervullen. Aluminium kan
door zuurstof tetraëdrisch of octaëdrisch worden gecoördineerd. Al3+ is in staat om de plaats
van Si4+ in de tetraëdrische holten van de silicaatstructuur in te nemen, maar het kan ook
tweewaardige ionen zoals Mg2+ en Fe2+ in de grotere octaëdrische holten vervangen.
Bovendien kunnen in één kristalstructuur zowel tetraëdrisch als octaëdrisch gecoördineerde
Al3+-ionen voorkomen. Als driewaardig aluminiumatoom de positie inneemt van een
vierwaardig siliciumatoom wordt de totale lading één ladingseenheid kleiner. Daarom is er
een bijkomend kation zoals H+ of Na+ vereist voor elk aluminiumatoom dat een
siliciumatoom vervangt. De ladingscompensatie van twee Al/Si-substituties kan ook door een
tweewaardig ion zoals Ca2+ worden gecompenseerd. Aluminiumhoudende silicaten met
(Si,Al)-subsitutie worden aluminosilicaten genoemd. Voorbeelden zijn de zeolieten,
veldspaten en veldspaatvervangers. De substitutiegraad van Si door Al kan de verhouding 1:1
niet overschrijden, d.w.z. dat een overgang van aluminosilicaten naar aluminaten niet
voorkomt. Silicaten waarbij Al3+ enkel in de octaëdrische holten voorkomt en waarbij dus
geen vervanging van Si4+ door Al3+ in de tetraëdrische holten plaats vindt, noemt men
aluminiumsilicaten (en geen aluminosilicaten). Aan de samenstelling van een aantal silicaten
nemen ook bijkomende anionen deel, zoals O2-, [OH]-, F-, Cl-, [CO3]2- en andere.
3.8 Chemische formules van anorganische kristalverbindingen
De gebruikelijke chemische formules zoals NaCl of CaF2 geven de stoechiometrische
verhoudingen weer van de atomen die aan de bouw van de kristalstructuur deelnemen. Als er
echter mengkristallen voorkomen, dan worden de elementen die elkaar substitueren tussen
ronde haakjes gezet en gescheiden door een komma. De elementen worden volgens dalende
116
hoeveelheden gerangschikt. Zo stelt Ag(Cl,Br) een chloorhoudend zilverbromide voor, terwijl
Ag(Br,Cl) staat voor een broomhoudend zilverchloride. (Zn,Fe)S stelt een ijzerhoudend
zinksulfide voor.
Om de coördinatiegetallen in de formules tot uitdrukking te brengen, kan men aan de
gebruikelijke chemische formules de coördinatiegetallen als een superscript tussen vierkante
haakjes rechts boven het element schrijven. Voorbeelden zijn Zn[4]S[4] voor sfaleriet, Na[6]Cl[6]
voor haliet, Ca[6][CO3] voor calciet, Ca[9][CO3] voor aragoniet, Mg2[6][SiO4] voor forsteriet
(olivijn), Ca3[8]Al2[6][SiO4]3 voor granaat, ... Deze notatie wordt vooral gebruikt voor
aluminiumsilicaten en aluminosilicaten om aan de duiden of Al3+ zich in een tetraëdrische
holte of in een octaëdrische holte bevindt. Een voorbeeld is sillimaniet, Al[6]Al[4][O⏐SiO4].
Voor verbindingen met complexe anionen bestaat er een bijzondere schrijfwijze:
Au[12]Bv[8]Cw[6][(O,OH,F)x⏐(RO3)y]⋅nH2O
voor carbonaten, nitraten en boraten,
Au[12]Bv[8]Cw[6][(O,OH,F)x⏐(RO4)y]⋅nH2O voor sulfaten, fosfaten en silicaten.
Hierbij bevinden de kationen zich voor de vierkante haakjes, en geordend volgens afnemende
ionstraal. Tussen de vierkante haakjes staat het anionisch gedeelte, waarbij een verticale lijn
de complexvreemde anionen scheiden van het eigenlijke complexe anion, dat rechts van de
verticale lijn staat. Beschouw als voorbeeld de formule van titaniet, Ca[7]Ti[6][O⏐SiO4].
Hieruit volgt dat het van de SiO4-tetraëder gescheiden O2--anion enkel aan de Ca2+- en Ti4+kationen is gebonden, terwijl het O2--anion van de SiO4-tetraëder zowel aan de genoemde
kationen als aan silicium is gebonden. Daarom worden de complexvreemde anionen dichter
bij de kationen geschreven dan het complexe anion.
Door
1
2
∞, ∞
,
3
∞
voor de formule te schrijven kan men aanduiden dat de structuur bestaat
uit resp. oneindige ketens, een laagstructuur of uit een driedimensioneel netwerk. Na de
formule kan dan eventueel ook de symmetrie van het kristalrooster worden weergegeven: c
(kubisch), h (hexagonaal), t (tetragonaal), trig (trigonaal), or (orthorhombisch), m
(monoklien), tr (triklien). Zo luidt de kristalchemische formule voor natriumchloride:
3
[6] [6]
∞ Na Cl c.
Hieruit kan men afleiden dat elke Na+-ion omgeven is door zes Cl--ionen en elk
Cl--ion omgeven is door zes Na+-ionen. De structuur strekt zich in alle richtingen van de
ruimte uit en de symmetrie is kubisch. Op eenzelfde manier kan men voor sfaleriet schrijven
3
[4] [4]
∞ Zn S c
en voor cadmium(II)chloride
2
[2]
[6]
∞ Cd Cl2 h.
117
Hoofstuk 4:
Niet-stoechiometrische verbindingen
4.1 Thermodynamica en onvolmaakte kristallen
In een volmaakt kristal zijn alle atomen in rust op hun correcte roosterplaatsen. In
principe kan een perfect kristal enkel bij het absolute nulpunt worden verkregen. Atomen
vibreren en deze vibraties kunnen beschouwd worden als een vorm van kristaldefect. Een
aantal atomen zal zich echter niet meer op de oorspronkelijke roosterplaats bevinden.
Het is reeds een paar eeuwen bekend dat er gekleurde variëteiten bestaan van mineralen
die normaal kleurloos zijn, zoals haliet (steenzout, NaCl), fluoriet (CaF2), korund (Al2O3) en
kwarts (SiO2). Denken we maar aan de korundvariëteiten robijn en saffier, en aan de
kwartsvariëteiten rookkwarts en amethist. De kleur van deze mineralen wordt door
roosterfouten of defecten veroorzaakt. Roosterfouten kunnen beschouwd worden als
onvolkomenheden van de structuur of samenstelling. Roosterfouten zijn van belang omdat ze
eigenschappen zoals de mechanische sterkte, elektrische geleidbaarheid en chemische
reactiviteit beïnvloeden. In sommige kristallen zoals in silicium, kwarts of diamant met een
zeer hoge zuiverheid, is het aantal roosterfouten zeer klein (<< 1%). In sommige kristallen
zijn echter zoveel roosterfouten aanwezig dat men zich kan afvragen om de defecten zelf niet
als een fundamenteel onderdeel van de kristalstructuur moeten beschouwd worden, eerder dan
een vorm van onvolkomenheid in een anders ideale structuur.
De belangrijkste reden voor het voorkomen van roosterfouten is dat een zekere
concentratie aan defecten leidt tot een verlaging van de Gibbs vrije energie ΔG van het
systeem:
ΔG = ΔH - TΔS
(4.1)
Op het eerste gezicht lijkt het invoeren van lege plaatsen in een ionische rooster energetisch
onvoordelig: het verwijderen van ionen vermindert immers het aantal elektrostatische
interacties en bijgevolg ook de roosterenthalpie. Dit betekent dat ΔH positiever wordt, wat
minder gunstig is. Het invoeren van lege plaatsen in het kristalrooster leidt echter ook tot een
toename van de entropie ΔS van het systeem. De toename van de entropie is aanzienlijk
omwille van het groot aantal posities waar de roosterfout kan optreden. De entropie is immers
ook een maat voor het aantal mogelijkheden om wanorde in een systeem in te voeren. Het
effect is het meest uitgesproken als het kristalrooster sterk wordt verstoord door het invoeren
van de eerste roosterfouten. Naarmate dat de concentratie aan roosterfouten toeneemt, wordt
118
de verstoring van het kristalrooster door de bijkomende roosterfouten minder uitgesproken.
Hoe meer roosterfouten men invoert, hoe meer dit de enthalpie ΔH doet stijgen. Het totale
effect wordt getoond in Figuur 3.1, waaruit duidelijk wordt dat ΔG een minimum bereikt voor
een bepaald aantal roosterfouten. Aangezien de term TΔS temperatuursafhankelijk is, is de
concentratie aan roosterfouten ook temperatuursafhankelijk.
Figuur 4.1. De variatie van de enthalpie en de entropie van een kristal als het aantal
roosterfouten toeneemt. De resulterende Gibbs vrije energie, ΔG = ΔH - TΔS,
bereikt een minimum bij een concentratie verschillend van nul, zodat de vorming van
roosterfouten spontaan verloopt.
4.2 Typen van roosterfouten
Aangezien een kristalrooster een geometrisch concept is, ligt het voor de hand om
roosterfouten geometrisch in te delen. Zo kent men nuldimensionale roosterfouten
(puntvormig), ééndimensionale roosterfouten (lijnvormig) en tweedimensionale roosterfouten
(vlakvormig).
Roostertrillingen
kunnen
als
driedimensionale
roosterfouten
worden
beschouwen. We bespreken in deze sectie enkel de puntvormige en de lijnvormige
roosterfouten.
119
Figuur 4.2. Tweedimensionale voorstelling van een eenvoudige kristallijne vaste stof, die
enkele van de mogelijke puntvormige roosterfouten laat zien.
Figuur 4.3.Tweedimensionale voorstelling van een ionische vaste stof, die een aantal
mogelijke puntvormige roosterfouten vertoont. Kationen worden voorgesteld door kleine
bolletjes, anionen door grote.
120
Een puntvormige roosterfout is een zeer gelokaliseerde onderbreking van de regelmaat
van het kristalrooster. Een puntvormige roosterfout doet zich in de regel voor tengevolge van
de afwezigheid van een matrixatoom (een atoom dat er in een volmaakt kristal wel zou zijn),
de aanwezigheid van een atoom van een verontreiniging of van een matrixatoom op een
“verkeerde” plaats (een in een volmaakt kristal niet bezette plaats). De meest voorkomende
puntvormige roosterfouten zijn geïllustreerd in Figuur 4.2. Enkel van degene die in een
ionenkristal kunnen voorkomen zijn in Figuur 4.3 getoond. De afwezigheid van een atoom uit
een normaliter bezette positie wordt een vacante plaats of vacature genoemd. Een vreemd
atoom dat zich op de plaats van een matrixatoom bevindt, noemt men een substitutioneel
atoom en een vreemd atoom in een tussenruimte tussen matrixatomen (interstice) noemt men
een interstitieel atoom. Behalve deze zijn er nog puntvormige roosterfouten van complexere
aard, bijvoorbeeld groepen van meerdere vacatures, zoals dubbele en drievoudige vacatures.
In ionenkristallen kan een klein kation zijn plaats verlaten, daardoor een kationvacature
scheppend en zichzelf “oplossen” in de tussenruimte van de structuur: zo een stel bij elkaar
behorende defecten (een kationvacature geassocieerd met een interstitieel kation) noemt men
een Frenkel-defect. Wanneer een kationvacature samengaat met een anionvacature in plaats
van met een interstitieel kation, wordt het paar (een anion- met bijbehorende kationvacature)
een Schottky-defect genoemd. Over het algemeen is het aannemelijker dat zich Schottkydefecten vormen dan Frenkel-defecten, omdat er weinig structuren zijn die voldoende grote
tussenruimtes bevatten om zonder aanzienlijke spanningen kationen op te nemen. Niettemin
kunnen in een betrekkelijk open structuur als die van fluoriet (CaF2), waar grote tussenruimtes
zijn, kationen heel gemakkelijk in tussenruimten oplossen en Frenkel-defecten veroorzaken.
Figuur 4.4. 2D-voorstelling van een Schottky-defect in NaCl.
121
Het Schottky-defect is het voornaamste type van puntdefect in alkalihaliden. De
stoechiometrie van een kristal wordt niet veranderd door de aanwezigheid van een Schottkydefect omdat er gelijke aantallen onbezette plaatsen zijn op de kation- en anionplaatsen. Een
voorbeeld van een Schottky-defect in NaCl is in Figuur 4.4 weergegeven). Een vacante
anionplaats in NaCl heeft een netto lading + 1 omdat de vacante plaats is omringd door zes
natriumionen, elke met een gedeeltelijk niet-gecompenseerde lading. Men kan ook zeggen dat
een onbezette anionplaats een lading +1 heeft omdat na plaatsen van een anion met lading –1
op deze plaats de lokale elektroneutraliteit wordt hersteld. De concentratie aan Schottkydefecten is sterk afhankelijk van het type verbinding. De concentratie is heel laag in de
alkalihaliden en bedraagt slechts ongeveer 10-12 mol/dm3 bij 130 °C. Deze concentratie komt
overeen met ongeveer één defect per 1014 formule-eenheden. Aan de andere kant hebben
sommige oxiden, sulfiden en hydriden van d-blokmetalen vaak een zeer grote concentratie
aan Schottky-defecten. Een extreem voorbeeld is de hoge-temperatuurvorm van TiO, waar de
concentratie aan Schottky-defecten ongeveer 10 mol/dm3 bedraagt, wat overeenkomt met één
defect per 10 formule-eenheden. Naast in NaCl, komen Schottky-defecten vaak voor in MgO,
CaO, LiF, LiCl, LiBr, LiI en KCl.
Een Frenkel-defect in zilverchloride waarbij een Ag+-ion is verplaatst naar een
interstitiële plaats, wordt in Figuur 4.5 getoond. Hierbij bezetten in AgCl, dat de NaClstructuur heeft, een klein aantal Ag+-ionen tetraëdrische sites. De Ag+-ionen zijn tetraëdrisch
omgeven door vier Cl--ionen, maar ook op dezelfde afstand door vier Ag+-ionen. Het
interstitieel Ag+-ion bevindt zich bijgevolg op een acht-gecoördineerde site met vier Ag+ionen en vier Cl--ionen als naaste buren. Er is waarschijnlijk een zekere covalente interactie
tussen het interstitieel Ag+-ion en de vier Cl--ionen die zorgt voor een stabilisatie van het
Frenkel-defect. Daarom komen in AgCl Frenkel-defecten voor en geen Schottky-defecten.
Het harde Na+-ion heeft daarentegen een meer uitgesproken kationisch karakter en voelt zich
niet comfortabel in een site die tetraëdrisch door vier andere Na+-ionen wordt omringd.
Frenkel-defecten komen omzeggens niet voor in NaCl. Er zijn twee types van Frenkeldefecten mogelijk: kation-Frenkel-defecten en anion-Frenkel-defecten. Het anion-Frenkeldefect komt niet zoveel voor omdat anionen groter zijn dan kationen en zich daarom
moeilijker doorheen het kristalrooster naar een interstitiële plaats kan bewegen. In
verbindingen met de fluoriet-structuur (CaF2) zijn anion-Frenkel-defecten wel algemeen. De
anionen hebben in dit geval een kleinere lading. Bovendien bezetten de anionen tetraëdrische
holten, terwijl de grote octaëdrische holten leeg zijn. Naast in AgCl en CaF2, komen Frenkeldefecten vaak voor in SrF2, UO2, ZrO2, CeO2 en ThO2.
122
Figuur 4.5. (a) 2D-voorstelling van een Frenkel-defect in AgCl. (b) Interstitiële site die
tetraëdrische coördinatie door Ag en Cl toont.
Naast structurele onvolkomenheden kunnen andere puntvormige roosterfouten die niet
structureel van aard zijn, veroorzaakt worden door een kristal te bestralen met
hoogenergetische straling of een bepaalde chemische behandeling te laten ondergaan. Een
voorbeeld van zulke een puntvormige roosterfout is een kleurcentrum. Dit is een algemene
term voor roosterfouten die verantwoordelijk zijn voor de verandering van de infraroodzichtbaar- en ultravioletabsorptie-eigenschappen van vaste stoffen die werden bestraald met
hoogenergetische straling of die een chemische behandeling ondergingen. Een welbekend
voorbeeld van een kleurcentrum is een F-centrum (afgeleid van het Duitse woord voor
kleurcentrum, Farbenzentrum). Het bestaat uit een elektron gevangen in een anionvacature
(Figuur 4.6). Zo kan een F-centrum worden gevormd door NaCl te verwarmen in
natriumdamp. Het NaCl wordt een beetje niet-stoechiometrisch door de opname van natriumatomen ter vorming van de verbinding Na1+δCl (δ << 1). Deze verbinding heeft een
geelgroene kleur. Het proces moet de absorptie van natrium-atomen omvatten. De natriumatomen ioniseren op het kristaloppervlak. De gevormde Na+-ionen blijven op het oppervlak
achter, maar de gevormde elektronen diffunderen in het kristal waarbij ze anionvacatures
tegenkomen en deze gaan bezetten. Om de elektrische neutraliteit te bewaren, moet een gelijk
aan Cl--ionen naar de oppervlake diffunderen. De elektronen die in de anion-site worden
ingevangen zijn een klassiek voorbeeld van een “deeltje in een doos”. Een reeks van
energieniveaus is beschikbaar voor het elektron in deze doos en de energie vereist om het
elektron van de grondtoestand naar een aangeslagen toestand te brengen komt overeen met de
energie van straling uit het zichtbare gedeelte van het elektromagnetische spectrum. De
waargenomen kleur is afhankelijk van het gastheerkristal en niet van de elektronenbron.
Bijgevolg heeft NaCl verhit in kaliumdamp dezelfde geelachtige kleur als NaCl verhit in
natriumdamp, terwijl KCl verhit in kaliumdamp violet is. Een andere methode om een
123
kleurcentrum te vormen is door het materiaal bloot te stellen aan een X-stralenbundel die de
elektronen door ionisatie in de anionvacatures vrijzet. Zo wordt poedervormig NaCl
groengeel door bestraling met X-stralen. De oorzaak van de kleur zijn eveneens gevangen
elektronen, maar deze keer kunnen ze niet afkomstig zijn van een niet-stoechiometrische
overmaat van natrium. De elektronen zijn waarschijnlijk afkomstig van de ionisatie van een
aantal chloride-ionen.
Figuur 4.6. F-centrum in NaCl.
Puntvormige roosterfouten zijn moeilijk direct waar te nemen met X-stralendiffractie
omdat kleine willekeurige afwijkingen van de periodiciteit niet worden opgemerkt. Soms
tonen spectroscopische metingen de aanwezigheid aan van een ion op een ongewone positie
of van een elektron gevangen in een bepaalde site in het rooster. De aanwezigheid van een
groot aantal vacante plaatsen of extra roosteratomen kan afgeleid worden uit verschillen
tussen de berekende en de experimentele massadichtheid. Ook de elektrische geleidbaarheid
kan wijzen op de aanwezigheid van defecten. Moderne elektronenmicroscopie kan zulk een
hoge resolutie bereiken dat defecten direct kunnen waargenomen worden. Kleurcentra kunnen
waargenomen
worden
met
behulp
van
optische
spectroscopie
(UV-VIS
absorptiespectroscopie) of met behulp van elektronspinresonantie (ESR of EPR).
Schottky-defecten en Frenkeldefecten zijn intrinsieke roosterfouten, omdat ze voorkomen
in zuivere verbindingen. Deze roosterfouten hebben geen invloed op de stoechiometrie van de
verbinding. Men spreekt daarom ook wel over stoechiometrische roosterfouten. Ook
kleurcentra die door bestraling met hoogenergetische straling (zoals X-stralen) worden
geïnduceerd, zijn ook intrinsieke roosterfouten. Bij de F-centra verandert de stoechiometrie
van de verbinding echter wel. Zulke roosterfouten die niet-stoechiometrisch zijn en waarbij de
samenstelling van het kristal verandert, noemt men extrinsieke roosterfouten. Onzuiverheden
in kristallen zijn ook voorbeelden van extrinsieke roosterfouten. Extrinsieke roosterfouten zijn
onvermijdelijk omdat het onmogelijk is om een perfecte zuiverheid te hebben in een kristal
124
van een redelijke grootte. Extrinsieke defecten kunnen ook intentioneel worden ingevoerd
door een kristal met een atoom of ion te doteren. Extrinsieke roosterfouten zijn essentieel in
de niet-stoechiometrische verbindingen die hieronder worden besproken.
Lijnvormige roosterfouten worden vaak dislocaties genoemd. Twee veelvoorkomende
typen van dislocaties zijn randdislocaties (Figuur 4.7) en schroefdislocaties (Figuur 4.8). Een
randdislocatie is een extra halfvlak van atomen in het kristalrooster. Het is een vlak van
atomen dat slechts gedeeltelijk door de kristalstructuur doorloopt. Op de plaats waar dit vlak
in de kristalstructuur stopt heeft men een sterke vervorming van het kristalrooster. De
eindpunten van dit vlak vormen een lijn doorheen het kristal, waardoor men van een
lijnvormige roosterfout spreekt. Bij een schroefdislocatie zijn de roostervlakken zodanig
vervormd dat ze als het ware een spiraaltrap rond de dislocatielijn beschrijven. De spoed van
de spiraal bedraagt soms slechts enkele transitie-eenheden (dus van enkele tot ca. 20 Å), maar
de spoed kan ook verscheidene honderden Å bedragen.
Figuur 4.7. Randdislocatie.
Figuur 4.8. Schroefdislocatie.
125
4.3 Niet-stoechiometrische verbindingen
Een niet-stoechiometrische verbinding is een verbinding met een variabele samenstelling,
maar waarbij de belangrijkste structurele kenmerken behouden blijven. Bij nietstoechiometrische verbindingen kunnen de verhoudingen tussen de atomen in de brutoformule
niet door gehele getallen worden weergegeven. Het lijkt daarom dat voor deze verbindingen
de wet van de constante massaverhoudingen (wet van Proust) niet geldig is. Vaak zijn nietstoechiometrische verbindingen vaste stoffen die willekeurige puntvormige roosterfouten
bevatten, zodat er een gebrek aan één van de samenstellende elementen optreedt. Een andere
benaming voor niet-stoechiometrische verbindingen is bertolliden, genoemd naar de Franse
chemicus Claude Louis Bertholet (1748-1822) die veronderstelde dat verbindingen met een
constante samenstelling eerder uitzonderingen dan regel waren. Verbindingen met een
constante samenstelling worden daarentegen daltoniden genoemd, naar de Britse chemicus
John Dalton (1766-1844).
Niet-stoechiometrie komt vaak voor bij oxiden van transitiemetalen, omwille van de
mogelijkheid van het transitiemetaalion om in verschillende oxidatietoestanden voor te
komen. Een zeer grondig onderzochte niet-stoechiometrische verbinding is wüstiet. De ideale
stoechiometrische formule van wüstiet is FeO, maar de werkelijke samenstelling is dichter bij
Fe0,95O. Het niet-stoechiometrisch gedrag wordt veroorzaakt door ontbreken van een aantal
Fe2+-ionen. Wüstiet is bijgevolg een niet-stoechiometrische verbinding met kationvacatures.
De samenstelling van een niet-stoechiometrische verbinding kan meestal over een eng
concentratiegebied continu worden gevarieerd. Daarom wordt de formule van wüstiet ook
geschreven als Fe1-xO, waarbij x een klein getal is, om de afwijking van de ideale formule
weer te geven. In het gegeven voorbeeld x = 0,05, maar x kan variëren tussen 0,89 en 0,96.
Wüstiet heeft de NaCl-structuur. Ook de niet-stoechiometrische wüstietverbindingen hebben
dezelfde structuur. De kubische roosterparameter daalt wel geleidelijk, naarmate het
ijzergehalte afneemt. De wet van Vegard zegt dat de roostercelparameters van vele nietstoechiometrische verbindingen geleidelijk met de samenstelling variëren, maar dat de
symmetrie van het kristalrooster onveranderd blijft. Dit gedrag is afwijkend van dat van de
stoechiometrische verbindingen met een constante massaverhouding zoals Fe3O4 en Fe2O3,
die verschillende structuren hebben. Het verwijderen van een aantal Fe2+-ionen zou enkel
kationvacatures mogen geven, waarbij de overblijvende Fe2+-ionen de octaëdrische sites
blijven bezetten. Onderzoek heeft echter aangetoond dat er meer kationvacatures zijn op de
octaëdrische sites dan vereist is door de chemische formule van de verbinding. Een aantal
ijzerionen blijkt tetraëdrische sites te bezetten en heeft oxidatietoestand +3. De afstand tussen
de octaëdrische en tetraëdrische holten is echter te klein opdat beide plaatsen in het rooster
kunnen bezet zijn.
126
In transitiemetaalmonoxiden MO kan de niet-stoechiometrische samenstelling op
verschillende manieren worden verwezenlijkt. Ten eerste kunnen er kationvacatures optreden
en wordt de formule door M1-xO voorgesteld. Een voorbeeld is Fe1-xO. Voor elk ontbrekend
M2+ ion, worden omwille van het behoud van de elektroneutraliteit twee M2+ tot M3+
geoxideerd. Ten tweede kunnen anionvacatures optreden. De algemene formule is dan MO1-x.
Een voorbeeld is VO1-x. Voor elke O2--vacature worden 2 M2+-ionen tot M+ gereduceerd. Ten
derde kunnen er interstitiële kationen optreden. De algemene formule is M1+xO. Voor elk
interstitieel M2+-ion, worden twee M2+-ionen tot M+ gereduceerd. Een voorbeeld is Zn1+xO.
Ten vierde kunnen er interstitiële anionen voorkomen, waarbij de algemene formule MO1+x is.
Voor elke interstitieel O2--ion worden er twee M2+-ionen tot M3+ geoxideerd. UO2+x heeft
interstitiële O2--ionen.
4.4 Vaste oplossingen
Als twee vloeistoffen worden gemengd vormen ze één fase zonder fasegrenzen, op
voorwaarde dat ze vergelijkbare dichtheden en polariteiten hebben. De moleculen van dit
homogeen mengsel zijn tot op moleculair niveau met elkaar vermengd. Een staal van dit
mengsel bevat de twee vloeistoffen in de verhoudingen zoals ze aan elkaar werden
toegevoegd. Een vaste oplossing is vergelijkbaar met een vloeibare oplossing, maar het is
vaste stof. De toegevoegde atomen of ionen lossen als het ware in de basisstructuur op en
verdwijnen erin. Het is een vast homogeen mengsel van zuivere stoffen. Daarom worden een
kristallijne vaste oplossing ook wel mengkristal genoemd. Een vaste oplossing heeft evenals
de hierboven besproken niet-stoechiometrische verbindingen geen constante samenstelling.
Terwijl bij een niet-stoechiometrische verbinding de afwijking van de ideale stoechiometrie
eerder gering is, vertoont een vaste oplossing een grotere continue variatie van de
samenstelling zonder dat er een verandering in het structuurtype plaatsvindt. In veel gevallen
zijn alle mogelijke verhoudingen tussen de samenstellende componenten mogelijk zonder dat
er fasescheiding optreedt. Men onderscheidt twee typen van vaste oplossingen. Bij een
substitutionele vaste oplossing vervangen de toegevoegde atomen of ionen direct de atomen
en ionen in het kristalrooster van de basisstructuur. Bij een interstitiële vaste oplossing
worden de atomen of ionen geplaatst in de holten (interstices) van het kristalrooster van de
basisstructuur. Beide typen van vaste oplossingen beïnvloeden de eigenschappen van het
materiaal door de kristalstructuur te verstoren. Als een vaste stof wordt gedoteerd met een
onzuiverheid, treden ook substituties in het kristalrooster of bezetting van interstitiële plaatsen
op juist zoals bij vaste oplossingen. Men spreekt van een gedoteerde vaste stof als de
onzuiverheid laag is (max. 1%), terwijl bij vaste oplossingen het dotage-gehalte veel hoger is.
127
4.4.1 Substitutionele vaste oplossingen
Eén van de eenvoudigste typen van vaste oplossing houdt de vervanging in van ionen met
dezelfde lading als degene die reeds in het kristalrooster aanwezig zijn. Dit proces treedt in de
natuur op als α-alumina (korund) wordt gedoteerd met Cr3+ ter vorming van robijn. Al2O3 en
Cr2O3 hebben op hoge temperatuur dezelfde kristalstructuur en Cr3+ kan gemakkelijk Al3+
vervangen in het kristalrooster van Al2O3. De vaste oplossing wordt geformuleerd als (Al2xCrx)O3,
waarbij 0 ≤ x ≤ 2. Bij intermediaire x-waarden zijn Al3+ en Cr3+ willekeurig verdeeld
over de octaëdrische sites die door Al3+ in Al2O3 worden ingenomen. Terwijl een bepaalde
site Al3+ of Cr3+ moet bevatten, is de waarschijnlijkheid dat het een bepaald ion is, gerelateerd
met de samenstellingsparameter x. Als de structuur als een geheel wordt beschouwd en een
gemiddelde van de bezetting van alle sites wordt gemaakt, is het nuttig om te beschouwen
alsof elke site wordt bezet door een “gemiddeld kation” waarvan de eigenschappen zoals
atoomnummer, ionstraal, ... intermediair zijn tussen deze van Al3+ en Cr3+.
Terwijl het systeem Al2O3 ⎯ Cr2O3 een goed eenvoudig voorbeeld is van een
substitutionele vaste oplossing, toont het systeem ook op een zeer duidelijke manier aan hoe
de eigenschappen van een vaste stof kunnen veranderd worden door doteren of vorming van
een vaste oplossing. Zuiver kristallijn Al2O3 (korund) is een kleurloze vaste stof. Wanneer
kleine hoeveelheden (< 1%) van Al3+ door Cr3+ vervangen wordt, wordt de kleur helderrood.
Dit is de samenstelling van de edelsteen robijn en het was ook de samenstelling van het
materiaal waarin het principe van laser-werking voor het eerst werd gedemonstreerd
(robijnlaser). Het zeer uitgesproken effect van de kleine hoeveelheid onzuiverheid wordt nog
duidelijker als de Cr3+-concentratie wordt verhoogd. De vaste oplossingen krijgen dan de
groene kleur van Cr2O3.
Om een substitutionele vaste oplossing te kunnen vormen moet er aan een aantal
voorwaarden zijn voldaan. Deze voorwaarden die de vorming van vaste oplossingen dirigeren
worden de Hume-Rothery-regels genoemd, naar de Britse metallurg William Hume-Rothery
(1899-1966) die de samenstelling van legeringen heeft bestudeerd. De regels zijn:
(1) De ionen die elkaar vervangen moeten dezelfde lading hebben.
(2) De ionen of atomen die elkaar vervangen moeten een vrij gelijke grootte hebben.
(3) De twee eindleden moeten dezelfde kristalstructuur hebben.
(4) De twee elementen moeten een gelijkaardige elektronegativiteit (of elektropositiviteit)
hebben.
Hoe meer er van deze regels wordt afgeweken, des te minder de ionen of atomen elkaar in een
kristalstructuur kunnen uitwisselen.
128
Als de ionen die elkaar vervangen niet dezelfde lading hebben, moeten andere structurele
veranderingen die vacatures and interstices inhouden optreden om de elektrische neutraliteit
te behouden. In het geval van de vorming van legeringen heeft men gevonden dat de straal
van de metaalatomen die elkaar vervangen slechts een verschil van maximaal 15% mogen
vertonen opdat vaste stoffen over een vrij groot concentratiegebied vaste oplossingen kunnen
vormen. Voor vaste oplossingen in niet-metallische systemen is het toelaatbaar verschil in
ionstraal groter dan 15%, maar het is moeilijk om deze limiet te bepalen. De meting van
ionstralen is immers zeer moeilijk en bovendien is de vorming van vaste oplossingen sterk
temperatuursafhankelijk. Op basis van een oxide-ionstraal van 1,26 Å in een octaëdrische
omgeving, vindt men dat monovalente kationen de volgende ionstraal (in Å ) hebben: Li+,
0,88; Na+, 1,16; Ag+, 1,29; K+, 1,52; Rb+, 1,63; Cs+, 1,84. De stralen van de paren K/Rb en
Rb/Cs verschillen elk minder dan 15% van elkaar elkaar en zouten van deze paren vormen
vaak vaste oplossingen. Alhoewel het K+-ion ongeveer 30% groter is dan het Na+- ion,
vormen natrium- en kaliumzouten soms vaste oplossingen, zeker bij hoge temperaturen (bijv.
KCl en NaCl bij 600 °C). Het verschil in grootte tussen Li+ en K+ is echter te groot, en deze
ionen vervangen elkaar normaliter niet in vaste oplossingen. Het Ag+-ion heeft ongeveer
dezelfde grootte als hat Na+-ion. Bijgevolg vormen natriumzouten en zilverzouten
gemakkelijk vaste oplossingen. De ionstralen (in Å ) van een aantal tweewaardige ionen in
een octaëdrische omgeving zijn: Mg2+, 0,86; Ca2+, 1,14; Sr2+, 1,30; Ba2+, 1,50; Mn2+ 0,96
(high spin); Fe2+, 0,91 (high spin); Co2+, 0,88 (high spin); Ni2+, 0,84; Cu2+, 0,87; Zn2+, 0,89;
Cd2+, 1,09. Vaste oplossingen waarin Mn2+ en Zn2+ elkaar vervangen komen veel voor. Dit is
niet verwonderlijk omwille van de vrij gelijke ionstraal van Mn2+ en Zn2+. Mg2+ vormt ook
vaak vaste oplossingen waarin het Mn2+ of Zn2+ vervangt. Ca2+ is echter niet in staat om Mn2+,
Zn2+ of Mg2+ te vervangen omdat het 20-30% groter is. Gelijkaardige overwegingen zijn van
toepassing op de driewaardige ionen. Zo hebben Al3+, Ga3+, Fe3+ en Cr3+ een gelijkaardige
ionstraal (0,67-0,76Å) en zijn in staat om elkaar in kristalstructuren te vervangen. De
driewaardige lanthanide-ionen (driewaardige ionen van zeldzame aarden) hebben een
gelijkaardige grootte (0,99-1,20 Å) vandaar dan in ertsen van zeldzame aarden steeds een
mengsel van deze elementen voorkomt. Omdat Zr4+ (0,86 Å) en Hf 4 + (0,85 Å) omzeggens
dezelfde ionstraal hebben, spreekt het voor zich dat deze ionen gemakkelijk elkaar kunnen
vervangen in kristalstructuren en vaste oplossingen vormen. Deze vaste oplossingen zijn bij
allle temperaturen stabiel.
Bij hoge temperaturen is vorming van vaste oplossingen zeer algemeen, terwijl deze bij
lagere temperaturen meer beperkt is en aan striktere voorwaarden is onderworpen. Er is een
eenvoudige thermodynamische verklaring voor de temperatuursafhankelijkheid van de
vorming van vaste oplossingen. Een vaste oplossing is stabiel als het een lagere Gibbs vrije
energie heeft dan het overeenkomstige mengsel dat geen vaste oplossingen vormt. Zoals
129
hogerop in dit hoofdstuk besproken kan de vrije energie opgesplitst worden in een enthalpieen entropiebijdrage. We kunnen het effect van de vorming van een vaste oplossing op elk van
deze twee bijdragen beschouwen. De entropie-term begunstigt altijd de vorming van een vaste
oplossing omdat de entropie van een vaste oplossing, die bijvoorbeeld bestaat uit twee typen
van kationen die willekeurig zijn verdeeld over een roosterpunten van het kristalrooster, veel
groter is dan de entropie van het overeenkomstige mengsel van de twee stoechiometrische
eindleden. Of de enthalpie-term al dan niet de vorming van een vaste oplossing begunstigt, is
afhankelijk van de situatie. Als de vormingsenthalpie negatief is, dan wordt de vorming van
een vaste oplossing begunstigd door zowel de entropie- als de enthalpiebijdrage. De
gevormde vaste oplossing is dan bij alle temperaturen stabiel. Als de vormingsenthalpie
echter positief is, dan werken de entropie- en enthalpie-termen elkaar tegen. De enthalpie
domineert de vrije energie bij lage temperatuur en bij deze temperaturen zal er geen
uitgesproken vorming van vaste oplossingen optreden. De term TΔS neemt met stijgende
temperatuur toe. Bij een bepaalde temperatuur zal deze term de positieve enthalpie meer dan
compenseren. Bijgevolg worden vaste oplossingen bij hoge temperaturen begunstigd. Als de
ionen een sterk gelijkende ionstraal hebben is de mengenthalpie zeer klein en is de toename in
entropie de drijvende kracht voor de vorming van vaste oplossingen. Zulke vaste oplossingen
zijn bij alle temperaturen stabiel. Als de ionen 15-20 % in grootte verschillen, kunnen vaste
oplossingen zich op hoge temperatuur vormen, waar entropie-term in staat is om de positieve
enthalpie-term te compenseren. Als de ionen meer dan ongeveer 30% in grootte verschillen,
verwacht men niet dat vaste oplossingen zich vormen. Voor systemen die op hoge
temperatuur een vaste oplossing vormen, maar niet op lage temperatuur, treedt exsolutie op.
Bij exsolutie scheidt de vaste oplossing zich in twee discrete kristallijne fasen. De twee
gescheiden fasen kunnen microscopisch worden waargenomen. Een bekend voorbeeld van
exsolutie komt voor bij nikkelijzermeteorieten. Deze vertonen afscheiding van een nikkelrijke
fase (taeniet) en een ijzerrijke fase (kamaciet). Wanneer aan doorgezaagde en gepolijste
ijzernikkelmeteoriet met een oplossing van salpeterzuur in methanol wordt geëtst, wordt het
ijzerrijke kamaciet sterker geëtst dan het nikkelrijke taeniet. Hierbij ontstaat de zeer
karakteristieke Widmanstätten-figuren.
Een belangrijke factor die bepaalt op vaste oplossingen zich al dan niet vormen is de
kristalstructuur van de twee eindleden. In systemen die een vaste oplossing vormen over het
hele concentratiegebied, is het essentieel dat de eindleden isostructureel zijn (d.w.z. dat ze
dezelfde kristalstructuur hebben). Het omgekeerde is niet noodzakelijk het geval. Het is niet
zo dat als twee verbindingen isostructureel zijn, dat ze dan ook met elkaar vaste oplossingen
zullen vormen. LiF en CaO hebben beide de NaCl-structuur, maar ze vormen geen vaste
oplossingen.
130
Terwijl zich in gunstige gevallen een vaste oplossing kan vormen over het hele
concentratiegebied van de twee eindleden, zoals in het geval van het systeem Al2O3 ⎯ Cr2O3,
komt het veel vaker voor dat vaste oplossingen zich slechts vormen over een gedeeltelijk of
slechts een beperkt concentratiegebied tussen de eindleden. In zulke gevallen hoeven de
eindleden niet isostructureel te zijn. Bijvoorbeeld, Mg2SiO4 (forsteriet) en Zn2SiO4 (willemiet)
kunnen voor ongeveer 20% in elkaar oplossen ter vorming van een vaste oplossing, alhoewel
hun kristalstructuren verschillend zijn. In de vaste oplossingen van forsteriet met de formule
Mg2-xZnxSiO4 kunnen een aantal van de octaëdrisch gecoordineerde Mg2+-ionen door Zn2+
vervangen worden. In de vaste oplossingen van willemiet met de formule Zn2-xMgxSiO4
kunnen Mg2+-ionen gedeeltelijk de tetraëdrisch gecoördineerde Zn2+-ionen vervangen. Zulke
vaste oplossingen zijn mogelijk omdat Mg2+ en Zn2+ een gelijkaardige ionstraal hebben en
omdat ze beide in een octaëdrische of een tetraëdrische omgeving kunnen voorkomen.
Bepaalde ionen, vooral de transitiemetaalionen, hebben een sterke voorkeur voor een
bepaalde symmetrie of coördinatiegetal. Zo wordt Cr3+ bijna steeds in octaëdrische sites
gevonden, terwijl het Al3+-ion, dat bijna even groot is, zowel in tetraëdrische als in
octaëdrische sites kan voorkomen. Zulke voorkeur voor bepaalde sites sluit de vorming van
bepaalde vaste oplossingen uit. Bijvoorbeeld LiCrO2 bevat octaëdrische Cr3+-ionen en vormt
vaste oplossingen met de formule LiCr1-xAlxO2, waarbij Al3+ gedeeltelijk Cr3+ in de
octaëdrische sites vervangt. De omgekeerde vaste oplossing vormt zich niet, aangezien in
LiAlO2 de Al3+-ionen zich in een tetraëdrische omgeving bevinden en Cr3+ een sterke afkeer
voor een tetraëdrische coördinatie heeft. Verschillende types van legeringen zijn niets anders
dan substitutionele vaste oplossingen. Bijvoorbeeld, in messing vervangen koper en zink
elkaar over een breed bereik aan samenstellingen, met als algemene formule Cu1-xZn.
4.4.2 Interstitiële vaste oplossingen
Vele metalen vormen interstitiële vaste oplossingen waarbij kleine atomen zoals H, C, B,
N de interstitiële sites in de gastheermetaalstructuur bezetten. Het is bekend dat palladium
enorme hoeveelheden waterstofgas kan opsluiten ter vorming van een hydride die een
interstitiële vaste oplossing is. De algemene formule is PdHx: 0 ≤ x ≤ 0,7, waarbij de Hatomen de interstitiële sites bezetten in de metaalstructuur van palladium, die een
vlakgecentreerde kubische structuur is. Het is nog niet met zekerheid geweten of de H-atomen
de octaëdrische, dan wel de tetraëdrische sites bezetten. Bovendien lijkt het dat het type van
site dat wordt bezet van x afhankelijk is. Ook staal is een voorbeeld van een interstitiële vaste
oplossing. In deze legering bezetten koolstofatomen een klein gedeelte van de interstitïele
sites in het kristalrooster van α-Fe. In Figuur 4.9. wordt het verschil tussen substitutionele en
interstitiële vaste oplossingen getoond.
131
Figur 4.9. (a) Substitutionele vaste oplossing (b) Interstitiële vaste oplossing.
4.5 Glas
4.5.1 Chemische samenstelling en structuur van glas
Glas is een anorganisch materiaal dat vanuit de gesmolten toestand door afkoelen zonder
te kristalliseren in de vaste toestand overgaat. Een glas is een amorfe (of niet-kristallijne)
vaste stof. Het is een wijdverbreid misverstand dat glas een onderkoelde vloeistof is. Een
onderkoelde vloeistof gedraagt zich echter als een echte vloeistof, maar dit is niet het geval
voor glas. Men zou glas wel kunnen beschouwen als een bevroren onderkoelde vloeistof. Het
verschil in afkoelgedrag tussen een glas en een normale kristallijne stof is duidelijk wanneer
men de verandering van het volume van een hoeveelheid glas en kristallijn materiaal als
functie van de temperatuur beschouwt (Figuur 4.10). Als een vloeistof afkoelt, neemt in het
algemeen het volume af en de dichtheid wordt groter. Voor een gesmolten materiaal zal, als
de smelttemperatuur (stoltemperatuur, Tf,) wordt bereikt, de vloeistof stollen. Het
stollingsproces gaat gepaard met het sterk krimpen van het materiaal. Bij afkoelen tot
kamertemperatuur zal het volume steeds verder afnemen. Voor een gesmolten glas is het
132
afkoelingsgedrag anders. Bij de temperatuur waarbij het stollen van het materiaal zou moeten
plaatsvinden (de stollingstemperatuur) treedt er in de glassmelt geen kristallisatie op. De
glassmelt wordt een onderkoelde vloeistof en het volume verandert niet stapsgewijs, maar
blijft geleidelijk afnemen bij afkoelen volgens de vloeistoflijn. Tegelijkertijd neemt bij
afkoelen de viscositeit (stroperigheid) van het gesmolten glas toe. Bij een bepaalde
temperatuur verstart het glas en een niet-kristallijne vaste stof wordt gevormd. De atomen of
moleculen waaruit het glas is opgebouwd kunnen dan niet meer bewegen. De temperatuur
waarbij de glassmelt verstart is de glastransitietemperatuur, Tg. Als glas heel snel afkoelt,
wordt het invriezen van de glassmelt tot een niet-kristallijne stof uitgesteld en wordt het
volume van het glas groter dan van glas met exact dezelfde samenstelling maar dat heel
langzaam is afgekoeld (Figuur 4.11)
Figuur 4.10. Vergelijking van de volumeverandering als functie van de temperatuur voor
onderkoelde vloeistoffen en glas, vergeleken met een kristallijn materiaal. Tf is de
stoltemperatuur en Tg is de glastransitietemperatuur.
Figuur 4.11. Invloed van de afkoelsnelheid op het volume van een hoeveelheid glas.
133
De meeste in de glastechniek toegepaste glassoorten bevatten als hoofdbestanddeel
siliciumdioxide (SiO2). De eigenschappen van een bepaalde glassoort wordt hoofdzakelijk
bepaald door de aard en de hoeveelheid van de verschillende oxide-componenten, waaruit het
glas is samengesteld. De oxiden die het netwerk vormen dat noodzakelijk is voor glasvorming
worden netwerkvormers genoemd en ze worden gekarakteriseerd door een bindingenthalpie
voor kation-zuurstof binding van meer dan 335 kJ/mol. Naast SiO2, kunnen ook GeO2, B2O3,
P2O5, As2O5, Sb2O5 en V2O5 als netwerkvormer optreden.
Een glas wordt gemaakt door een smelt sneller af te koelen dan de kristallisatiesnelheid.
Zo verkrijgt men kwartsglas door snel afkoelen van gesmolten silica. Onder deze
omstandigheden heeft de vaste massa geen periodiciteit over lange afstanden. Op atomaire
schaal is in kwartsglas elk siliciumatoom tetraëdrisch door vier zuurstofatomen omgeven,
juist zoals in kristallijn kwarts. Het gebrek aan orde over lange afstanden in kwartsglas is te
wijten aan variaties in de Si-O-Si hoeken. In Figuur 4.12 wordt in twee dimensies getoond
hoe een goed-gedefineerde lokale coördinatie-eenheid kan behouden blijven, maar dat de
lange-afstandsorde verloren gaat door variatie van de bindingshoeken rond zuurstof.
Siliciumdioxide vormt gemakkelijk een glas omdat het driedimensionele netwerk van sterke
covalente Si-O-bindingen in de smelt niet gemakkelijk gebroken wordt en bij afkoeling niet
gemakkelijk opnieuw gevormd wordt. Het ontbreken van sterke directionele bindingen in
metalen en in eenvoudige ionische verbindingen zorgen ervoor dat het veel moeilijker is om
glazen van deze verbindingen te maken. Het concept dat de lokale coördinatiesfeer van het
glasvormende element behouden blijft, maar dat de bindingshoeken rond zuurstof variabel
zijn, werd oorspronkelijk in 1932 voorgesteld door de Amerikaanse kristallograaf W. H.
Zachariasen.
134
Figuur 4.12. Schematische voorstellling van (a) twee-dimensionaal kristal en
(b) twee-dimensionaal glas.
Kwartsglas dat uit zuiver SiO2 bestaat is een hoogsmeltende (smeltpunt boven 1700 °C),
corrosievaste,
maar
moeilijk
te
verwerken
glassoort.
Het
heeft
een
zeer
lage
uitzettingscoëfficiënt waardoor het snel verhit of afgekoeld kan worden zonder stuk te
springen. Het heeft bovendien een zeer grote doorlatendheid voor UV-straling. Kwartsglas is
de enige in de industrie gebruikte ééncomponentglassoort. Kwartsglas wordt toegepast in een
aantal hoogwaardige producten zoals speciale laboratoriumapparaten, halogeenlampen en
glasvezels. Omdat kwartsglas een zeer hoge glastransitietemperatuur heeft, kan het enkel bij
zeer hoge temperaturen verwerkt worden. Daarom wordt er gewoonlijk een netwerkwijziger
(of verzachter) zoals Na2O aan SiO2 toegevoegd. Deze netwerk-wijzigers verbreken enkele
van de Si-O-Si-bindingen en vervangen ze door eindstandige Si-O--bindingen die met het
kation geassocieerd zijn (Figuur 4.13). Deze consequente gedeeltelijke verbreking van het SiO-netwerk leidt tot de vorming van glazen die bij een lagere temperatuur zacht worden. Het
gevolg is dat de verwerkingstemperatuur aanzienlijk daalt. Daarentegen wordt de thermische
uitzettingscoëfficiënt verhoogd, waardoor de bestendigheid tegen thermische schokken daalt,
en ook de bestandheid tegen chemische corrosie daalt. Netwerkwijzigende oxiden zijn Li2O,
Na2O, K2O, MgO, CaO en La2O3. De netwerkwijzigende kationen nemen interstitiële plaatsen
in het netwerk in. Het netwerk kan uitzetten (bijv. met K+-ionen) of inkrimpen (bijv. door Li+ionen) afhankelijk van de grootte en het aantal van deze kationen. MgO reduceert de neiging
van het glas om uit te kristalliseren. Daarnaast zijn er aantal amfotere oxiden, zoals Al2O3,
135
PbO, ZnO en ZrO2, die afhankelijk van het glastype als netwerkvormer of als netwerkwijziger
kunnen optreden. Het toevoegen van Al2O3 verhoogt de chemische bestendigheid maar ook
het smeltpunt van het glas, waardoor het verwerkingsproces wordt bemoeilijkt.
Figuur 4.13. Invloed van een netwerkwijziger op de glasstructuur.
Het gewone glas dat in flessen en voor vensterglas wordt gebruikt heet natronkalkglas en
bevat Na2O en CaO als netwerkwijziger. Het wordt reeds bij 700 °C week en is daarom veel
gemakkelijker dan kwartsglas te verwerken. Wanneer een deel van SiO2 als netwerkvormer
door
B2O3
wordt
vervangen,
verkrijgt
men
borosilicaatglas
dat
een
lagere
uitzettingscoëfficiënt dan natronkalkglas heeft en bijgevolg minder gemakkelijk stukspringt
bij verhitting. Borosilicaatglazen hebben ook een grotere chemische stabiliteit dan
natronkalkglazen. Borosilicaatglas wordt toegepast in laboratoriumglaswerk en voor
ovenschalen. Een bekend type van borosilicaatglas is Pyrex®. Lood(II)oxide treedt in de
meeste gevallen op als een netwerkwijziger en in uitzonderlijke gevallen ook als
netwerkvormer. Toevoeging van lood aan glas leidt tot loodglazen met een laag smeltpunt.
Omwille van zijn hoge brekingsindex is loodglas geschikt om als kristalglas (loodkristal)
gebruikt te worden. De netwerkvormers en netwerkwijzigers vormen niet in alle
verhoudingen een glas. De verhoudingen van componenten die wel een glas kunnen vormen
kunnen in het geval van drie componenten in een ternair fasediagram worden weergegeven.
136
Als voorbeeld zijn in Figuur 4.14 de glasvormingsgebieden voor de systemen Na2O – CaO –
SiO2 en Na2O – B2O3 – SiO2 weergegeven. Een glas kan worden gevormd uit verschillende
combinaties van verschillende componten. Door de grote keuze aan toe te voegen
bestanddelen zij de eigenschappen van het materiaal glas over een breed gebied te variëren.
Dit is ook één van de redenen die geleid hebben tot een brede technologische inzetbaarheid
van glas. In Tabel 4.1 is de chemische samenstelling van een aantal belangrijke glassoorten
weergegeven. Ijzer(III)oxide (Fe2O3) is in een aantal glazen aanwezig en is meestal afkomstig
van onzuiverheden in de grondstoffen. Fe2O3 veroorzaakt groenkleuring van het glas. Als er
naast Fe2O3 ook nog zwavel aanwezig is in het glas, treedt er een sterke bruinkleuring op door
de vorming van colloïdale Fe-S deeltjes, zoals in glas voor bierflessen of voorraadpotten voor
chemicaliën. Daarnaast kan er doelbewust kleurende bestanddelen aan glas worden
toegevoegd. Zo kleurt CoO glas donkerblauw, CuO lichtblauw, Cr2O3 donker- tot
smaragdgroen, Nd2O3 lila, Er2O3 roze. Grote concentraties aan MnO2 kleuren onder
oxiderende omstandigheden glas amethistkleurig (paars). UO3 geeft aan glas een geelgroene
kleur. Uraniumglas fluoresceert intens groen bij bestraling met ultraviolette straling.
Nanodeeltjes van metallisch goud geven aan glas een zeer typische paarsrode kleur.
Nanodeeltjes van cadmiumsulfide of zilver kunnen glas geel kleuren. Een intens geel
gekleurd glas wordt verkregen door een mengsel van TiO2 en CeO2.
Tabel 4.1. Chemische samenstelling van een aantal voor de glastechniek meest belangrijke
glazen (in gewichtsprocenten).
SiO2
Na2O
K2O
MgO
CaO
B2O3
Al2O3
rest
- kleurloos
72,6
13,7
0,5
0,1
11,0
---
1,6
0,2SO3;0,1TiO2
- groen
72,0
15,1
---
2,1
8,4
---
1,1
0,4Fe2O3;
verpakkingsglas
0,2Cr2O3
- bruin
72,7
13,8
1,0
---
10,0
---
1,9
0,2Fe2O3; 0,3SO3
tafelglas
75,6
13,5
4,1
2,6
3,7
---
0,4
vensterglas
72,8
12,8
0,8
3,8
8,2
---
0,4
opaalglas
66,9
13,3
2,2
0,4
4,8
---
6,9
1,6BaO;6F
loodglas
54,9
0,2
12,9
---
---
---
---
32PbO
kwartsglas
100
---
---
---
---
---
---
pyrex
80,2
4,5
0,3
---
0,1
12,3
2,6
137
Figuur 4.14 Ternaire fasediagrammen waarin de glassamenstelling van floatglas en
borosilicaatglas staat weergegeven.
4.5.2 Glaseigenschappen
De meest in het oog springende eigenschappen die ertoe geleid hebben dat glas een zo
breed toepassingsgebied heeft gevonden is het smelt- en viscositeitsgedrag van dit materiaal.
De viscositeit van glas is sterk temperatuursafhankelijk. In Figuur 4.15 worden de viscositeit
van een aantal soorten glas met elkaar vergeleken. Bij de verwekingstemperatuur is de
viscositeit gedaald tot 107 Pa⋅s, en bij deze viscositeit wordt een glazen voorwerp duidelijk
vervormd onder zijn eigen gewicht. Het verwekingsgebied of glasverwerkingsgebied is het
temperatuursgebied waarbij de viscositeit tussen 103 Pa⋅s en 107 Pa⋅s gelegen is, en waarbij de
vormgeving kan plaatsvinden (Figuur 4.16). Als dit temperatuursinterval klein is (150-250
°C) dan spreken we van een kort glas, is dit gebied groot (> 300 °C) dan spreken we van een
lang glas. De viscositeit van een glas bij de temperatuur waarbij het uit de grondstoffen wordt
gesmolten bedraagt 3 tot 30 Pa⋅s. Glas heeft dus zelfs bij de hoge temperatuur in de smeltoven
een een relatief hoge viscositeit als we dit vergelijken met de viscositeit van andere
vloeistoffen (de viscositeit van water bij 20 °C bedraagt 0,001 Pa⋅s of 1 cP). Bij de
smelttemperatuur is het glas wel dunvloeibaar genoeg om homogeen en bellenvrij te kunnen
worden gesmolten. De reden voor de hoge viscositeit is te vinden in de manier waarop het
glas uit moleculen is opgebouwd (zie hogerop). De moleculaire eenheden zijn zelfs bij de
hoge smelttemperaturen veel sterker met elkaar verbonden dan de moleculen in andere
138
vloeistoffen. Als de temperatuur van een glassmelt vanaf zijn smelttemperatuur wordt
verlaagd, neemt de viscositeit toe volgens een curve zoals in Figuur 4.17 wordt getoond. Deze
curve geldt in het algemeen voor alle glassoorten. De viscositeit neemt toe omdat bij
temperatuursverlaging steeds meer sterke chemische bindingen tussen de moleculaire
eenheden in de glassmelt worden gevormd. Door deze sterke bindingen kunnen bij de
temperatuur waarbij het gesmolten glas zou moeten kristalliseren de moleculen hun vaste
plaats die ze in een kristalrooster innemen niet meer vinden. Het glas kristalliseert daarom
niet en de viscositeit neemt bij afkoelen steeds verder toe tot een viscositeit van 1012 Pa⋅s
(glastransitietemperatuur) wordt bereikt en alle moleculen “vastvriezen” en zich niet meer
kunnen verplaatsen. Dit gebeurt bij een temperatuur tussen 400 en 600 °C. Het nietkristallijne materiaal glas is dan gevormd.
Figuur 4.15. Viscositeit van vier verschillende typen glas als functie van de temperatuur. (1)
kwartsglas; (2) borosilicaatglas; (3) natronkalkglas; (4) laagsmeltend loodboraatglas.
Figuur 4.16. Optimale bewerkingstemperaturen van glas met bijbehorende viscositeiten.
139
Een zeer belangrijke eigenschap van de meeste industrieel gebruikte glazen is de
transparantie voor zichtbaar licht. Als een glas geen kleurgevende componenten bevat, of niet
door kristallisatie en/of fasescheiding wit en ondoorzichtig wordt, is het doorlatend voor het
zichtbare gedeelte van het elektromagnetisch spectrum (400-800 nm). Daarnaast is glas,
enigzins afhankelijk van de glassamenstelling, doorlaatbaar voor ultraviolette en infrarode
straling. De doorlaatbaarheid in het ultraviolette gedeelte van het spectrum hangt af van het
type glas, maar ook in sterke mate van de aanwezigheid van onzuiverheden zoals ijzer(III).
Kwartsglas is gekend voor zijn goede doorlaatbaarheid voor ultraviolette straling. Het wordt
daarom ook gebruikt voor cuvetten voor spectroscopische metingen en voor lenzen in
spectrofotometers. In het infrarode gebied zijn de meeste glazen transparant tot ongeveer
2500 nm.
Glas is een bros materiaal. Het is sterk als het op druk wordt belast, maar zwak onder
trekkrachten. Deze eigenschap bepaalt in sterke mate hoe een glas als constructiemateriaal
kan worden toepast: altijd onder drukspanning. De sterkte van glas is nauwelijks afhankelijk
van de glassamenstelling, maar is wel in sterke mate afhankelijk van de mate waarin het
glasoppervlak is beschadigd. De massadichtheid van de meeste industriële glassoorten is
tussen 2,2 en 2,6 g cm-3 gelegen, maar de massadichtheid van loodglas is aanzienlijk hoger.
De specieke weerstand van glas is tussen 1011 en 1012 Ω⋅cm gelegen en daalt sterk met
toenemende temperatuur. Op kamertemperatuur is kwartsglas een zeer goede isolator voor
elektriciteit. Het glasoppervlak, vooral dat van alkalirijke glassoorten in een vochtige
atmosfeer, vertoont oppervlaktegeleiding.
Glas is een materiaal dat over het algemeen niet doorlatend is voor zowel vloeistoffen als
gassen. Gassen die bestaan uit kleine atomen (helium) of moleculen (waterstof) zijn wel in
staat om te diffunderen door sommige glazen met een relatief open glasstructuur, zoals
kwartsglas en siliciumdioxiderijke borosilicaatglazen. Glazen zijn gekenmerkt door hun hoge
resistentie tegen bijna alle chemicaliën en solventen. Waterstoffluoride tast echter glas snel
aan door de vorming van H2SiF6. De weerstand tegen waterige zuren en basen is sterk
afhankelijk
van
de
glassamenstelling
en
de
temperatuur.
Borosilicaat-
en
aluminiumsilicaatglazen vertonen de beste zuurbestendigheid. Sterke basen (logen) tasten glas
aan door depolymerisatie van het silicaatnetwerk tot silicaat-anionen. De basebestendigheid
kan verhoogd worden door toevoeging van CaO, MgO, Al2O3, B2O3 en vooral ZrO2. Water is
ook in staat om glas aan te tasten. Dit is de oorzaak van verwering van glas. Als men
gedestilleerd water een tijdlang in een beker van natriumcalciumsilicaatglas laat staan, dan
bevat het water duidelijk meetbare hoeveelheden aan opgeloste natrium-, calcium- en
silicaationen.
Bij temperaturen hoger dan de glastransitietemperatuur (Tg) gedraagt glas zich als een
onderkoelde vloeistof. In het gebied tussen de glastransitietemperatuur en het smeltpunt kan
140
er kristallisatie optreden van bepaalde componenten in het glas. Dit kristallisatieproces is
uiterst storend, men name in glasproductieprocessen waarbij glas voor een vrij lange tijd op
temperaturen in het kristallisatiegebied moet worden gehouden. Het kristallisatieverschijnsel
kan echter ook onder gecontroleerde omstandigheden worden uitgevoerd bij de fabricatie van
glaskeramieken. De gecontroleerde kristallisatie gebeurt volgens een tweestapsproces:
kristalkiemvorming (nucleatie) en de feitelijke kristallisatie. Kristalkiemen kunnen in het glas
worden gevormd bij temperaturen die slechts een paar honderd graden hoger dan de
glastransitietemperatuur zijn. Als er voldoende kristalkiemen zijn gevormd wordt de
temperatuur tussen 100 en 300 °C verhoogd om de eigenlijke kristalgroei te laten plaats
vinden. Hoe meer kiemen er tijdens de kristalkiemvorming worden gevormd, hoe fijner
kristallijn het glas is. De kristalfractie in glaskeramieken varieert tussen 50 en 95%. Na de
gehele of gedeeltelijke kristallisatie van glas veranderen de eigenschappen van het materiaal
volledig. Als de kristallieten duidelijk kleiner zijn dan de golflengte van licht (ca. 50 nm) en
als de brekingsindex van de kristallen en de glasfase hetzelfde zijn, is het glaskeramiek helder
en transparant.
In tegenstelling tot gewone gesinterde keramische stoffen hebben
glaskeramieken geen poriën tussen de kristallen. Glaskeramieken zijn mechanisch veel sterker
dan glas. Een voordeel van glaskeramieken is hun grote temperatuursstabiliteit in vergelijking
met gewoon glas. Vooral lithium-aluminiumsilicaatglazen vormen glaskeramieken die zeer
bestendig tegen thermische schokken zijn. Ze worden gebruikt voor het vervaardigen van
werktuigen die tegen temperatuursschokken bestand moeten zijn en voor keramische
kookplaten.
4.5.3 Glasbereiding
De belangrijkste grondstof voor de bereiding van silicaatglazen is fijn wit zand (SiO2).
Ruwe materialen voor de netwerkwijzigers zijn kalksteen (CaCO3), dolomiet (CaMg(CO3)2),
natriumcarbonaat
(NaCO3,
watervrije
soda)
en
veldspaten
(natrium-,
kalium-
of
calciumaluminosilicaten). Boor wordt toegevoegd onder de vorm van boorzuur, borax of
andere boormineralen. Andere toevoegstoffen worden onder de vorm van een oxide of
carbonaat toegevoegd. Voor de glasbereiding wordt ook vaak fijnvermalen gerecycleerd glas
aan de batch toegevoegd (van 10 tot 50%). Voor opsmelten wordt het mengsel grondig
gemengd en gehomogeniseerd. Op dit stadium worden de kleurgevende bestanddelen
toegevoegd. Soms worden enkele percenten water aan het mengsel toegevoegd om
stofvorming te vermijden en om het homogeniseren te vereenvoudigen.
De belangrijkste onzuiverheden in de grondstoffen zijn nikkel, koper, vanadium,
mangaan, chroom en vooral ijzer. Deze onzuiverheden zorgen voor ongewenste verkleuring
van het eindproduct, waarbij de kleur afhankelijk is van de redoxomstandigheden (oxiderende
141
of reducerende omstandigheden) waarbij de glassmelt wordt verwerkt. Voor glas van een
hoge kwaliteit, zoals optisch glas voor lenzen, moeten zeer zuivere startproducten worden
gebruikt. Het maximaal toelaatbaar Fe2O3-gehalte voor een UV-doorlatend glas is slechts
0,004% en voor vensterglas is dit 0,1%. Het ontkleuren van glas is belangrijk om goedkope
startmaterialen met een zeker ijzergehalte te kunnen gebruiken. Chemisch ontkleuren wordt
gebruikt voor Fe2O3-gehalten lager dan 0,1%. Bij dit proces worden oxiderende reagentia die
zuurstof leveren bij hoge temperatuur (KNO3, CeO2, Sb2O3) toegevoegd aan het glasmengsel.
Deze oxideren Fe2+ tot Fe3+, dat een minder intense kleur geeft. Bij hogere ijzeroxide gehalten
kan de groengele kleur van het glas fysisch gecompenseerd worden door gekleurde additieven
zoals MnO2. Het gekleurde additief heeft de complementaire kleur van de ijzeronzuiverheden,
waardoor het glas een lichtgrijze schijn verkrijgt.
Glas is een reactieproduct. Het ontstaat als een mengsel van de samenstellende
componenten op hoge temperatuur wordt gebracht en een versmelting optreedt. Bij
opwarming smelten eerst de componenten met een laag smeltpunt die als flux optreden en die
reageren met de componenten met een hoog smeltpunt. Er worden vloeibare fasen gevormd
en er treden ook vastestofreacties op. Zo reageert natriumcarbonaat met het oppervlak van de
zandkorrels bij 550 °C:
Na2CO3 + SiO2 → Na2SiO3 + CO2
Vanaf 700 °C verloopt de reactie verder:
Na2SiO3 + SiO2 → Na2Si2O5
Tenslotte vanaf 780 °C wordt de vloeibare fase van een eutectische samenstelling
3Na2Si2O5⋅SiO2 gevormd. De andere carbonaten en de andere hoogsmeltende componenten
zoals de veldspaten reageren op een gelijkaardige manier. De batch wordt aan elkaar gesinterd
en gaat tussen 1200 en 1600 °C in een gesmolten toestand over. De verkregen smelt is echter
nog niet homogeen en bevat nog vele gasbellen afkomstig van het ingesloten water en lucht in
de batch en van de gassen die tijdens de chemische reacties worden gevormd (CO2, O2). De
hoeveelheid gassen die gevormd worden in een typische natriumkalkglas bedraagt ongeveer
10% van de batch. De gasbellen worden verwijderd door het klaren van de glassmelt. Dit
wordt verkregen door toevoegen van additieven die de gassen uit de smelt uitdrijven en de
gasbellen naar de oppervlakte doen opstijgen. Hiervoor is vooral natriumsulfaat, dat ontbindt
ter vorming van SO3, heel geschikt. De glassmelt wordt gedurende verscheidene uren op een
142
hoge temperatuur gehouden tijdens dit zuiveringsproces. Tenslotte wordt de gassmelt
afgekoeld tot een temperatuur waarbij deze tot de eindproducten kan worden verwerkt.
4.5.4 Glasbewerking
Vlakglas wordt bereid met behulp van een uniek industrieel proces dat het floatproces
(drijfproces) wordt genoemd (Figuur 4.17). Daarom duidt men het vlakglas dat als vensterglas
wordt gebruikt ook wel aan met de term drijfglas of floatglas. Deze techniek waarmee kant en
klaar vlakglas met een perfect oppervlak kan worden gemaakt dateert uit 1957 en werd
ontwikkeld door Sir Alastair Pilkington. De essentie van dit procédé ligt in het drijven van
glas op een bad van vloeibaar tin. Dit tinbad met een lengte van ca. 50 meter is een onderdeel
van het gehele treksysteem. Na het tinbad passeert het glas achtereenvolgens de koelsectie, de
controle, de snij-eenheid en de afneem-eenheid. De treksnelheid en de transportsnelheid
worden geregeld via de looprollen waar het glas op ligt. Het glas wordt op breedte en dikte
gebracht op het tinbad. Het vloeibare glas wordt uit de glasoven via een brede lipsteen continu
op het tinbad gegoten. De temperatuur van het glas is direct na het verlaten van de glasoven
ca. 1100 °C. Bij deze temperatuur stroomt het glas breed uit en wordt eventueel met behulp
van koelers op ca. 900 °C gebracht. Door middel van zogenaamde toprollers wordt het glas
tot de gewenste breedte gestrekt en door regeling van de treksnelheid wordt de vereiste dikte
verkregen. Er wordt een doorlopend glaslint gevormd. Aan het einde van het tinbad is de
glastemperatuur tot ca. 600 °C gedaald. Hier wordt het glas van het tinbad gelicht. Het tinbad
bevindt zich in een van de buitenlucht afgesloten ruimte die continu met een mengsel van
stikstof en waterstof wordt gespoeld om oxidatie van het tin te vermijden. Het glas moet
geleidelijk afkoelen, onder voortdurende controle. Zou het glas te snel afkoelen, kunnen er
spanningsverschillen in het glas ontstaan, waardoor het kan breken. Na afgekoeld te zijn, kan
het glas op maat worden gesneden. Glasplaten worden veelal op een vaste handelsmaat (6 ×
3,21 meter) gesneden. Mechanische armen met zuignappen plaatsen het gesneden glas op
laadbokken,
waarna
het
klaar
is
voor
verzending.
Het
floatproces
heeft
een
productiecapaciteit van ongeveer 1000 ton/dag. Het is een volcontinu proces.
143
Figuur 4.17. Floatglasinstallatie.
Het persen van glas is een interessante en veelzijdige techniek voor het maken van
eenvoudig en goedkoop glaswerk zoals schalen, vazen, drinkbekers en siervoorwerpen. Het
gereedschap is relatief eenvoudig. Het bestaat in essentie uit drie delen: de persvorm, de
persstempel en de persring (Figuur 4.18). Deze laatste zorgt voor een afdichting tussen de
persvorm en de stempel, zodat het glas tijdens het persen alzijdig onder druk wordt gebracht,
waardoor een nauwkeurige vormoverdracht wordt gewaarborgd.
Figuur 4.18. Het persproces.
Glazen flessen worden met behulp van een blaasmachine vervaardigd. Een portie
gesmolten glas wordt in een voorvorm opgevangen en daar tot een preform omgevormd. De
preform ontstaat door het inblazen of inpersen van een holte in de glasdruppel. Hierbij
ontstaat tegelijkertijd de mond van de fles. De preform wordt daarna overgezet naar de
144
volblaasvorm, waar door uitblazen van de preform de fles ontstaat. Naargelang de preform
door inblazen van lucht of door middel van een persstempel wordt gevormd, spreekt men over
resp. de blaas-blaastechniek en de pers-blaastechniek (Figuur 4.19). De blaas-blaastechniek
wordt toegepast voor het vervaardigen van producten met een nauwe hals (flessen), terwijl de
pers-blaastechniek eerder wordt gebruikt voor het maken van producten met een wijde hals
(configuurpotten en dergelijke).
Figuur 4.19. Het blazen van glazen flessen. Bovenaan wordt het blaas-blaassysteem
getoond en onderaan het pers-blaassysteem.
145
Hoofdstuk 5:
Interpretatie van fasediagrammen
Fasediagrammen geven in grafische vorm de temperatuursgebieden (of drukgebieden) en
samenstellingen weer over dewelke fasen of mengsels van fasen stabiel zijn onder de
voorwaarde van thermodynamisch evenwicht. Ze bevatten bijgevolg informatie over de
samenstelling
van
verbindingen
en
mengkristallen,
over
fasetransities
en
over
smelttemperaturen. Fasediagrammen worden soms ook evenwichtsdiagrammen genoemd. De
faseregel van Gibbs is de fundamentele regel waarop fasediagrammen zijn gebaseerd. De
afleiding van de faseregel wordt hier niet gegeven, evenmin worden de thermodynamische
aspecten van de vrije energieën van fasen, vaste oplossingen en mengsels gedetailleerd
besproken. Hiervoor verwijzen we naar de cursus Chemische Thermodynamica. We staan
vooral stil bij de interpretatie en praktische aspecten van fasediagrammen.
5.1 Faseregel, fasen, componenten, vrijheidsgraden en evenwicht
De faseregel van Gibbs wordt gegeven door de vergelijking:
P+F=C+2
(5.1)
F=C-P+2
(5.2)
of
Hierbij is P het aantal fasen dat bij evenwicht aanwezig is, C is het aantal componenten dat
noodzakelijk is om het systeem te beschrijven en F is het aantal vrijheidsgraden of
onafhankelijke veranderlijken. De faseregel zegt dus dat het aantal vrijheidsgraden van een in
evenwicht verkerend systeem ⎯ dit is het aantal onafhankelijke veranderingen dat men in een
systeem kan aanbrengen zonder dat er fasen verdwijnen of nieuwe fasen worden gevormd ⎯
twee meer bedraagt dan het verschil tussen het aantal componenten en het aantal fasen in het
systeem. Hoe groter het aantal componenten van het systeem is, hoe meer grootheden
(temperatuur, dampdruk, samenstelling van de fasen) er vrij te kiezen zijn. Hoe meer fasen
men daarentegen naast elkaar in een systeem wil aantreffen, hoe meer bindend de
voorwaarden zijn waaraan het geheel moet voldoen.
146
Het woord fase is afgeleid van het Griekse woord voor “verschijning”. Een fase is een
toestand van de materie die geheel gelijkmatig is, niet alleen wat betreft de chemische
samenstelling, maar ook in zijn fysische toestand. Dit is de definitie zoals ze door de
Amerikaan Josiah Willard Gibbs (1839-1903) rond 1876 werd geformuleerd. Fasen zijn
homogene delen van een systeem, die door grensvlakken van elkaar zijn gescheiden en
daarom (in principe) langs mechanische weg uit het systeem kunnen worden afgezonderd. Zo
vormt bijv. een mengsel van water en ethanol, dat zich in een geheel gevuld een afgesloten
vat bevindt, een homogeen systeem, dat uit één fase bestaat. Water en ijs, met daarboven
waterdamp, vormen daarentegen een heterogeen systeem van drie fasen. In een willekeurig
stelsel kan nooit meer dan één gasfase voorkomen, terwijl het aantal daarin voorkomende
vaste fasen zeer groot kan zijn. Wat de vloeibare fasen betreft, deze blijken slechts in een
beperkt aantal naast elkaar te kunnen voorkomen. Een stelsel met vier vloeibare fasen behoort
reeds tot de hoge uitzonderingen. Dit komt omdat één-fase vloeibare oplossingen zich veel
gemakkelijker en over een groter samenstellingsgebied vormen dan één-fase vaste
oplossingen.
Het verschil tussen verschillende kristallijne fasen is gewoonlijk gemakkelijk
waarneembaar. Zo zijn in een fysisch mengsel van zwavelpoeder en ijzerpoeder de twee fasen
zonder problemen visueel onderscheidbaar. Het verschil tussen kristallijne fasen die
opgebouwd zijn uit dezelfde elementen, maar in verschillende verhoudingen is meestal ook
duidelijk waarneembaar. Zo vormen de magnesiumsilicaatmineralen enstatiet, MgSiO3, en
forsteriet, Mg2SiO4, verschillende fasen. Ze hebben een verschillende samenstelling, structuur
en eigenschappen. In het geval van vaste stoffen is het ook mogelijk om verschillende
kristallijne fasen met eenzelfde chemische samenstelling te hebben. Dit verschijnsel staat als
polymorfie (of als allotropie in het geval van chemische elementen) bekend. Als voorbeeld
kunnen we het mineraal calciet dat een trigonale kristalstructuur heeft en het mineraal
aragoniet dat een orthorhombische kristalstructuur heeft, maar die beide bestaan uit
calciumcarbonaat (CaCO3), vermelden. Een complicerende factor bij het klasseren van vaste
fasen is het voorkomen van vaste oplossingen (of mengkristallen): een vaste oplossing is een
enkelvoudige fase met een variabele samenstelling (zie Hoofdstuk 4). α-Al2O3 en Cr2O3
hebben dezelfde kristalstructuur (korundstructuur) en vormen een continue reeks van vaste
oplossingen bij hoge temperatuur. Een mengsel van Al2O3 en Cr2O3 kan op hoge temperatuur
reageren ter vorming van één enkele, homogene fase waarvan de structuur dezelfde is als deze
van de Al2O3 en Cr2O3 eindleden, maar waarbij de kationsites willekeurig door Cr3+ of Al3+
bezet zijn.
147
Het aantal componenten van een systeem is het aantal samenstellende bestanddelen die
een onafhankelijke variatie in de verschillende fasen kunnen ondergaan. Een alternatieve
beschrijving is dat het aantal componenten het kleinste aantal onafhankelijke bestanddelen is
waaruit men het gehele systeem met al zijn fasen kan opbouwen. Dit kan het beste aan de
hand van een aantal voorbeelden begrepen worden:
(a) Lost men NaCl op in water, dan ontstaat een systeem met twee componenten, daar
zowel water als NaCl onmisbaar zijn voor de opbouw van het systeem.
(b) Lost men willekeurige hoeveelheden NaCl en KNO3 samen in water op, dan stelt er
zich het volgende evenwicht in:
NaCl + KNO3 ' KCl + NaNO3
Deze vergelijking zou misschien tot de foutieve veronderstelling kunnen leiden dat er
in het verkregen systeem vijf componenten aanwezig zijn: H2O, NaCl, KNO3, KCl en
NaNO3. Dit systeem bevat echter slechts drie componenten. Het is immers geheel op
te bouwen uit water, NaCl en KNO3. Als we echter nog wat extra KCl of NaNO3 aan
de oplossing zouden toevoegen, dan bestaat het systeem uit vier componenten, want
uitgaande van de drie componenten NaCl, KNO3 en water zijn alleen zulke systemen
te verkrijgen waarin equimolaire hoeveelheden KCl en NaNO3 aanwezig zijn. Het is
natuurlijk duidelijk dat extra toevoegingen van NaCl en KNO3 geen invloed op het
aantal componenten hebben.
(c) Alle calciumsilicaten kunnen beschouwd worden als opgebouwd uit CaO en SiO2 in
variërende verhoudingen. Het is bijgevolg een twee-componentensysteem (binair
systeem), alhoewel er drie elementen, Ca, Si, O aanwezig zijn.
(d) De samenstelling “FeO” maakt deel uit van een twee-componentensysteem, ijzerzuurstof, omdat wüstiet in feite een niet-stoechiometrische, ijzer-deficiënte fase Fe1xO
is, veroorzaakt door de aanwezigheid van een beetje Fe3+. De bulkcomponent
“FeO” bevat een mengsel van twee fasen in evenwicht, zijnde Fe1-xO en Fe.
Het aantal vrijheidsgraden is het aantal onafhankelijke veranderlijken gekozen uit de
temperatuur, dampdruk en samenstelling van de fasen, d.w.z. het is het aantal variabelen dat
moet gespecifieerd worden opdat het systeem volledig gedefinieerd zou zijn. Dit kan ook het
beste aan de hand van een aantal voorbeelden geïllustreerd worden.
148
(a) Een systeem bestaande uit kokend water, d.w.z. water en stoom in evenwicht met
elkaar, hebben geen samenstellingsvariabele, omdat zowel water als stoom bestaan uit
moleculen met een welbepaalde vaste samenstelling, zijnde H2O. Om het systeem te
definiëren is het voldoende om enkel de stoomdruk te specifiëren, omdat dan het
kookpunt automatisch vastligt (en omgekeerd). Toepassing van de faseregel op dit
systeem geeft:
P + F = C + 2; C = 1 (i.e. H2O), P = 2 (damp en vloeistof)
Bijgevolg F = 1 (temperatuur of druk, maar niet beide)
Op zeeniveau kookt water bij 100 °C, maar in Mexico City (2285 meter boven
zeeniveau) bedraagt de atmosfeerdruk slechts 580 mbar en kookt water bij 92 °C. Het
systeem water-stoom is univariant, omdat slechts één vrijheidsgraad, ofwel druk
ofwel temperatuur, vereist is om het systeem bij evenwicht volledig te beschrijven.
Men moet er wel op wijzen dat de relatieve verhoudingen van water en stoom niet
door de faseregel worden gegeven. Zolang er voldoende stoom aanwezig is om de
evenwichtsdruk te behouden, is het volume van de damp onbelangrijk.
(b) Een vaste oplossing in het systeem Al2O3 ⎯ Cr2O3 heeft één samenstellingsvariabele,
omdat de Al2O3:Cr2O3 verhouding kan gevarieerd worden zodanig dat eenzelfde
homogene fase wordt verkregen. De temperatuur van deze één-fase vaste oplossing
kan ook gevarieerd worden. Er zijn bijgevolg twee vrijheidsgraden nodig om de vaste
oplossing volledig te karakteriseren, namelijk de samenstelling en de temperatuur.
Voor vele vaste stoffen met een hoog smeltpunt, is de dampspanning van de vaste stof en
zelfs deze van de vloeistof verwaarloosbaar in vergelijking met de atmosfeerdruk. De
dampfase is effectief onbestaande en moet bijgevolg niet beschouwd worden als een
mogelijke
variabele bij het werken op atmosfeerdruk. Zulke systemen worden
gecondenseerde systemen genoemd. De faseregel wordt aangepast zodat de gecondenseerde
faseregel wordt verkregen:
P+F=C+1
(5.3)
Bij gebruik van fasediagrammen is het belangrijk te verstaan wat met evenwicht wordt
bedoeld. Een systeem in evenwicht is in de laagste mogelijke energietoestand die in
overeenstemming is met de opgelegde voorwaarden. Het heeft geen neiging om spontaan te
veranderen; het is een stabiel systeem. Men kan beschouwen dat deze toestand op de bodem
van een vrije-energieput ligt (Figuur 5.1). Een systeem dat niet in evenwicht is is bezig te
149
veranderen of heeft de neiging om te veranderen. Het probleem bij de bepaling of het
evenwicht al dan niet werd bereikt is dat er minima in de vrije energie kunnen voorkomen, die
echter niet zo diep zijn als de evenwichtsput (dit zijn de zgn. lokale minima). Een systeem dat
zich in een lokaal minimum bevindt, noemt men een metastabiel systeem. Een metastabiel
systeem lijkt in evenwicht te zijn, maar is niet in de laagste energietoestand. Er kan een zeer
hoge energiebarrière betrokken zijn bij de overgan van een metastabiele naar de stabiele
toestand, en in vele omstandigheden is deze energiebarrière onoverkomelijk hoog. Een
voorbeeld is de metastabiliteit van diamant t.o.v. grafiet op kamertemperatuur. De
energiebarrière of activeringsenergie voor de reactie diamant → grafiet is zo hoog dat diamant
eens het gevormd wordt kinetisch stabiel, maar thermodynamisch onstabiel is. De
thermodynamisch betekenis van de term onstabiel is ook in Figuur 4.1 weergegeven. Als een
bal op de heuveltop wordt geplaatst, is de geringste beweging voldoende om de bal langs elk
van de twee kanten omlaag te laten rollen. Er is geen activeringsenergie betrokken bij de
overgang van een onstabiele naar een metastabiele of stabiele toestand. Voorbeelden van
onstabiel evenwicht zijn moeilijk te vinden, omwille van de onstabiliteit.
Figuur 5.1. Schematisch voorstelling van een metastabiele, onstabiele toestand en stabiele
toestand.
5.2 Unaire systemen
De onafhankelijke variabelen in een unair systeem (één-componentsysteem) zijn de
temperatuur en de dampdruk. De samenstelling kan hier geen onafhankelijke variabele zijn,
omdat er slechts één zuivere vaste stof aanwezig is en dus de samenstelling vast ligt. Uit de
faseregel volgt, P + F = C + 2 = 3. Het systeem is bivariant (F = 2) als er één fase aanwezig
is, univariant (F = 1) als er twee fasen aanwezig zijn en nonvariant (F = 0) als er drie fasen
150
aanwezig zijn. Vier fasen kunnen niet met elkaar in evenwicht zijn in een ééncomponentsysteem, omdat F niet negatief kan zijn.
Figuur 5.2. Schematische voorstelling van vast-vloeistof-dampevenwichten.
Tb is het normale kookpunt.
In Figuur 5.2 worden schematisch de vast-vloeistof-damp-evenwichten gegeven voor een
één-componentsysteem waarbij op de cartesische assen de onafhankelijke variabelen, de
temperatuur en de druk, staan uitgezet. Zulk een diagram wordt een P-T-fasediagram
genoemd. Alhoewel in dit P-T-diagram de temperatuur op de X-as is uitgezet en de druk op
de Y-as, vindt men in de literatuur ook veel fasediagrammen met de omgekeerde conventie.
De druk in fasediagrammen wordt vaak uitgedrukt in kilobar (kbar). Eén kilobar is gelijk aan
1000 bar en 1 bar is gelijk aan 0,9869 atmosfeer. De drie fasen zijn een vaste fase, een
vloeistoffase en een dampfase. Elke fase neemt een zeker gebied of veld in het fasediagram
in, aangezien F = 2 als P = 1 (zowel de druk als de temperatuur zijn vereist om om een punt
in één van deze gebieden te beschrijven). Zulk een veld of gebied noemt men ook wel
evenwichtsvlak. Elk één-fasegebied is gescheiden van naburige één-fasegebieden door
univariante curven (P = 2, en bijgevolg F = 1). Als op deze curven één van de variabelen,
bijv. de druk, wordt vastgehouden, dan is de waarde van de andere variabele, de temperatuur,
éénduidig bepaald. De lijn OA geeft de variatie van de dampspanning van de vloeistof als
functie van de temperatuur weer en stopt bij het kritische punt, waar vloeistof en damp
identiek zijn (niet getoond in de figuur). Deze lijn stelt de voorwaarden (van temperatuur en
151
druk) voor, waarbij de vloeistof en damp met elkaar in evenwicht zijn. Ze wordt verzadigde
dampspanningslijn of kooklijn genoemd. De lijn OB stel de variatie van de dampspanning van
de vaste stof als functie van de temperatuur, en ze loopt door tot het absolute nulpunt. Deze
lijn stelt de voorwaarden (van temperatuur en druk) voor, waarbij de vaste stof en damp met
elkaar in evenwicht zijn. Ze wordt sublimatielijn genoemd. De lijn OC geeft de temperaturen
en drukken weer waarbij de vaste stof en vloeistof met elkaar in evenwicht zijn. Met andere
woorden, OC geeft de invloed van de druk op het smeltpunt weer. De lijn OC wordt smeltlijn
genoemd. De helling van de smeltlijn hangt af van het feit of een druktoename het smeltpunt
verhoogt of verlaagt, d.w.z. of het specifiek volume van de vloeistof groter of kleiner is dan
dat van de vaste stof. In Figuur 4.2 helt naar links, wat betekent dat het smeltpunt daalt als de
druk stijgt. Dit is bijvoorbeeld het geval voor water en bismut. Er dient wel op gewezen te
worden dat in de figuur de helling van OC voor de duidelijkheid overdreven werd. De invloed
van de druk op het smeltpunt is zeer beperkt. De smeltlijn, kooklijn en sublimatielijn komen
samen in één punt O, dat het tripelpunt wordt genoemd en waarbij de vaste stof, de vloeistof
en de damp met elkaar in evenwicht zijn. De lijnen OA, OB en OC geven de voorwaarden
weer waarbij twee van de drie fasen naast elkaar kunnen bestaan, en alle drie fasen zijn met
elkaar in evenwicht waar de drie lijnen elkaar snijden. Aangezien de drie lijnen slechts één
snijpunt kunnen hebben, kan er slechts één tripelpunt zijn. De faseregel van Gibbs leert dat
een systeem met één component en drie fasen geen vrijheidsgraden meer heeft (F = 0,
nonvariant systeem). Als er drie fasen met elkaar in evenwicht voorkomen, ligt de
temperatuur en druk vast. Het normale smeltpunt van een vaste stof is de temperatuur waarbij
de vaste stof en vloeistof met elkaar in evenwicht zijn bij atmosfeerdruk. Bij het tripelpunt is
de druk echter gelijk aan de evenwichtsdampspanning van het systeem, waardoor de
temperatuur van het tripelpunt een weinig verschilt van het normale smeltpunt van de stof. Zo
bedraagt het normale smeltpunt van water 273,15 K (0 °C), terwijl het tripelpunt van water
ligt bij 273,16 K (0,01 °C) bij een druk van 611,73 Pascal (ca. 0,006 atmosfeer).
Uit een fasediagram zoals in Figuur 5.2 weergegeven kan heel wat informatie worden
afgeleid. Als de druk in een systeem bestaande uit vloeistof en damp in evenwicht met elkaar
constant wordt gehouden en als tegelijkertijd de temperatuur wordt verhoogd, dan is het
duidelijk dat alle vloeistof tenslotte in dampfase wordt omgezet. Zulk een verandering wordt
met de lijn lm in Figuur 5.2 weergegeven. Bij m is nog enkel damp aanwezig. Als de
temperatuur constant wordt gehouden en als tegelijkertijd de druk wordt verminderd, van l tot
n, zal volledige verdamping van de vloeistof optreden. Als een vast-dampsysteem,
voorgesteld door een punt op lijn OB, aan gelijkaardige veranderingen wordt onderworpen,
zal de vaste stof in damp worden omgezet. Het is bijgevolg duidelijk dat elk punt in het
gebied beneden de lijnen OB en OA enkel damp voorstelt. Als we terugkeren naar het
152
vloeistof-dampevenwicht bij l, kan men afleiden dat een verandering van de omstandigheden
naar o, d.w.z. een constante temperatuur en toename van de druk, of naar p, d.w.z. een
constante druk en afname van de temperatuur, zal leiden tot het verdwijnen van damp, zodat
enkel vloeistof aanwezig is. Toepassing van dezelfde redenering op punten van de lijnen OB
en OC, toont aan dat enkel vloeistof kan bestaan in het gebied tussen de lijnen OA en OC, en
dat enkel vaste stof kan voorkomen onder de voorwaarden van druk en temperatuur die
weergegeven wordt door het gebied tussen de lijnen OB en OC. Enkelvoudige fasen zijn
stabiel in de verschillende gebieden of velden getoond in Figuur 5.2. Twee gebieden komen
samen op een lijn, die de voorwaarden weergeeft waarbij twee fasen met elkaar in evenwicht
kunnen voorkomen. De drie lijnen komen samen in het tripelpunt.
Het is soms mogelijk om de kooklijn tot voorbij het tripelpunt door te trekken, zoals
weergegeven door lijn OA’ in Figuur 5.2. Dit gebeurt als de vloeistof onderkoeld is, d.w.z.
afgekoeld tot beneden het smeltpunt, zonder dat er stolling plaatsvindt. Het vloeistofdampsysteem is op lijn OA’ in een toestand van metastabiel evenwicht. Een metastabiel
systeem zal een spontane verandering ondergaan door toevoeging van de stabiele fase. Zo zal
toevoeging van een kleine hoeveelheid vaste stof aan een vloeistof-dampsysteem op lijn OA’
leiden tot het onmiddellijk vast worden van de gehele vloeistof. De temperatuur en druk
zullen dan weergegeven worden door een punt op lijn OB. Uit Figuur 5.2 kan ook afgeleid
worden dat de metastabiele toestand een hogere dampspanning heeft dan het stabiele systeem
bij dezelfde temperatuur. De realiseerbare metastabiele curve OA’ is meestal slechts kort,
omdat uiteindelijk een punt wordt bereikt waarbij het systeem spontaan naar een stabiele
toestand overgaat. Oververhitting van een vaste stof tot boven het tripelpunt treedt nooit op.
Daarom eindigt de sublimatielijn definitief bij het tripelpunt.
Stoffen die in meer dan drie fasen kunnen voorkomen zijn stoffen die het verschijnsel van
allotropie of polymorfie vertonen. Hierbij treden in de vaste fase verschillende modificaties
op. Allotropie is de naam die gewoonlijk gebruikt wordt als het verschijnsel bij een element
optreedt. Polymorfie is het analoge verschijnsel bij verbindingen. We kunnen hierbij twee
gevallen onderscheiden:
(1) Enantiotropie: Elk der modificaties heeft een eigen bestendigheidsgebied of
stabiliteitsgebeid. Er is een reële transitietemperatuur.
Voorbeelden:
orthorombische zwavel ' monokliene zwavel
stabiel als
T < 95,6 °C
T > 95,6 °C
153
grijs tin ' wit tin
stabiel als
T < 13,2 °C
rood HgI2
stabiel als
T > 13,2 °C
' geel HgI2
T < 126 °C
T > 126 °C
In het geval van enantiotropie kan de overgang van de ene modificatie naar de andere bij een
bepaalde temperatuur in beide richtingen plaats vinden. Het transitiepunt is de enige
temperatuur waarbij de twee vaste stoffen in evenwicht naast elkaar kunnen bestaan bij een
gegeven druk. Elke afwijking van deze temperatuur leidt tot de volledige transformatie van de
ene vorm in de andere, waarbij de ene vorm stabiel is boven het transitiepunt en de andere
beneden.
(2) Monotropie: Bij iedere temperatuur is één der modificaties stabiel, de andere metastabiel.
Er is geen reëel transitiepunt. De fasetransitie kan slechts in één richting plaats vinden,
namelijk van de metastabiele naar de stabiele vorm.
Voorbeelden:
witte fosfor ' rode fosfor
stabiel
metastabiel
grafiet ' diamant
stabiel
metastabiel
We zullen de fasediagrammen van zwavel, water en siliciumdioxide in detail bespreken.
5.2.2 Fasediagram van zwavel
Het volledige P-T-fasediagram van zwavel wordt in Figuur 5.3 getoond. Zwavel komt in
twee stabiele kristallijne vormen voor: een (ortho)rhombische vorm (α-S) en een monokliene
vorm (β-S). De structuur van de (ortho)rhombische α-zwavel bestaat uit ringvormige S8moleculen met een conformatie die lijkt op deze van een kroonether (zie ook Figuur 3.1). Bij
de overgang van rhombische naar monokliene zwavel blijven de S8-moleculen bestaan, maar
154
de pakking van de moleculen in de kristalstructuur wijzigt zich. Monokliene zwavel heeft een
kleinere massadichtheid (d = 1,94-2,01 g cm-3) dan rhombische zwavel (d = 2,069 g cm-3). AB
is de damplijn van rhombische zwavel. BC is de damplijn voor monokliene zwavel. De
rhombische vorm is onder zijn eigen dampspanning stabiel beneden 95,5 °C en de
monokliene vorm boven deze temperatuur. Het transitiepunt is door punt B in het fasediagram
aangeduid. Het smeltpunt van rhombische zwavel is 114,5 °C en dat van monokliene zwavel
119,25 °C. Het smeltpunt van rhombische zwavel kan enkel worden bereikt als zwavel snel
wordt verhit, zodat de (trage) transitie naar monokliene zwavel wordt onderdrukt. De lijn BF
toont het effect van de druk op het transitiepunt en lijn CF het effect van de druk op het
smeltpunt van de monokliene vorm. M.a.w. lijn BF geeft de voorwaarden voor evenwicht
tussen de rhombische en de monokliene vorm, terwijl lijn CF de voorwaarden voor evenwicht
tussen de monokliene vorm en de vloeistof weergeeft. De metastabiele lijn EF, die in de
stabiele lijn FG wordt verder gezet, toont de invloed van de druk op het smeltpunt van de
rhombische vorm. De vloeistof en rhombische zwavel zijn in evenwicht volgens de lijn EFG.
Het fasediagram toont vier tripelpunten. De drie fasen die hierbij in evenwicht staan zijn:
B: rhombisch, monoklien en damp
C: monoklien, vloeistof en damp
E: rhombisch, vloeistof en damp
F, rhombisch, monoklien en vloeistof
Men dient op te merken dat terwijl monokliene zwavel enkel echt stabiel is in gebied BFC, er
onder bepaalde omstandigheden toch rhombische zwavel kan voorkomen in BEF, vloeistof in
CEF en damp in BEC. Dit zijn echter allemaal metastabiele toestanden.
Het tripelpunt F (151 °C, 1290 atm) is van speciaal belang. Het ontstaat omdat zowel het
transitiepunt als het smeltpunt van monokliene zwavel toenemen met stijgende drukken, maar
de toename van het smeltpunt is meer uitgesproken dan het transitiepunt. Het
stabiliteitsgebied van monokliene zwavel wordt afgegrensd door de lijnen BF, CF en BC.
Monokliene zwavel kan niet buiten deze gebieden bestaan. Boven F kan zwavel in slechts één
stabiele vorm worden verkregen, namelijk rhombische zwavel. Als vloeibare zwavel bij een
initiële temperatuur en druk weergegeven door punt a in het fasediagram wordt afgekoeld,
zullen er bij overschrijden van de lijn FC monokliene kristallen worden afgezet. Bij snel
verder afkoelen naar kamertemperatuur kan de omzetting van monokliene naar rhombische
zwavel worden onderdrukt en kan men metastabiele monokliene zwavel bij kamertemperatuur
verkrijgen. Geleidelijk (over een tijdspanne van enkele weken) zal monokliene zwavel bij
kamertemperatuur wel in rhombische zwavel worden omgezet. Als de initiële druk en
temperatuur echter door punt b worden weergeven, dan zullen er bij afkoeling onder constante
155
druk rhombische zwavelkristallen worden gevormd bij overschrijden van lijn FG. Men
gelooft dat de grote kristallen van rhombische zwavel die in de natuur worden gevonden zijn
ontstaan door langzame uitkristallisatie van gesmolten zwavel bij hoge drukken. Bij verdere
verhitting van gesmolten zwavel blijft de dampdruk stijgen tot tenslotte het kritische punt D
wordt bereikt bij 1040 °C. Vloeibare zwavel is tussen 119 °C en 154 °C geel, dunvloeibaar en
bestaat uit S8-moleculen. Rond 160 °C breken de S8-moleculen open waarna door verknoping
van de losse uiteinden van de ketens hoogmoleculaire ketens en ringen (met 1000 of meer
zwavelatomen) ontstaan. De smelt wordt bruin en taaivloeibaar. In de buurt van het kookpunt
(444,6 °C) is de gesmolten zwavel opnieuw dunvloeibaar en bevat S8-deeltjes waarvan niet
bekend is of het hier over keten- of ringstructuren gaat. In de gasfase heeft men eerst S8moleculen (geel), bij 500 °C S6- en S4-moleculen (rood) en bij 650 °C S2-moleculen
(strogeel). Boven 1800 °C dissociëren de S2-moleculen tot S-atomen.
Figuur 5.3. Fasediagram van zwavel.
5.2.2 Fasediagram van water
Het fasediagram van water, getoond in Figuur 5.4, geeft voorbeelden van vast-vast en
vast-vloeibaar transities. Het tripelpunt wordt door O weergegeven. De stabiliteitsgebieden
van de verschillende kristallijne vormen van ijs worden met Romeinse cijfers aangeduid. Een
belangrijk verschil in vergelijking met het fasediagram van zwavel is dat bij water slechts één
polymorf, namelijk gewoon ijs (ijs I), stabiel is bij atmosfeerdruk. Gewoon ijs heeft een
156
hexagonale structuur die bestaat uit twee in elkaar gedrongen roosters met een hexagonale
dichtste bolstapeling. Het heeft een lagere dichtheid dan water (d = 0,917 g cm-3). Daarnaast
zijn er verschillende polymorfen bekend die stabiel zijn bij hoge drukken: ijs II tot ijs VII. Ijs
II is een geordende trigonale vorm van ijs (d = 1,17 g cm-3). Ijs III is tetragonaal en
wanordelijk (d = 1,14 g cm-3). Ijs I wordt bij een druk van ongeveer 2 kbar omgezet in ijs II of
ijs III. Onderaan de 5 km dikke ijskap van Antarctica bedraagt de druk slechts 0,5 kbar, zodat
een ijslaag op aarde nooit dik genoeg is om de omzetting naar een dichtere fase te doen plaats
vinden. Hogere drukken komen in de aardkorst voor, maar hier zijn de temperaturen ver
boven de transitietemperaturen van om het even welke type van ijs. Men neemt echter aan dat
de dichtere vormen van ijs wel voorkomen op ver van de zon gelegen planeten zoals Jupiter,
Saturnus, Uranus en Neptunus. Ijs V heeft een complexe wanordelijke monokliene structuur.
Ijs VI is tetragonaal en wanordelijk. Het bereikt heeft een grotere mate van verdichting omdat
het opgebouwd is uit twee in elkaar dringende netwerken van water moleculen (d = 1,31 g
cm-3); Ijs VII vormt kubische wanordelijke kristallen, waarbij een zeer dichte stapeling van de
watermoleculen plaatsvindt (d = 1,66 g cm-3). De structuur kan beschouwd worden als twee in
elkaar dringende netwerken met inwendig gecenterde kubische roosters. Theoretische
berekeningen tonen aan dat ijs VII tot een druk van tenminste 200 kbar stabiel is. Men kan
verwachten dat bij extreem hoge drukken de watermoleculen volledig in stukken breken en
dat er dicht gestapelde structuren met ionische bindingen worden gevormd. In alle bekende
kristalstructuren van de ijspolymorfen zijn de watermoleculen door waterstofbruggen
gebonden aan vier naburige watermoleculen. De H-O-H hoeken in de ijsfasen bedragen
ongeveer 107°, wat dichter bij de tetraëdrische hoek (109,47°) is dan in vloeibaar of
gasvormig water.
Uit het fasediagram kan afgeleid worden dat enkel ijs I, ijs III, ijs V, ijs VI en ijs VII in
evenwicht kunnen gebracht worden met vloeibaar water. Bij toename van de druk zullen dus
uit water bij afkoeling achtereenvolgens ijs I, ijs III, ijs V, ijs VI en ijs VII worden afgezet. Ijs
II is onder geen enkele voorwaarde van druk of temperatuur stabiel in de aanwezigheid van
vloeibaar water. Het kan dus enkel uitgaande van andere vormen van ijs, namelijk ijs I, ijs III
of ijs V, worden verkregen door een geschikte aanpassing van de temperatuur en druk. Ijs III
kan nooit bestaan in evenwicht met waterdamp. Merk op dat water bij zeer hoge drukken bij
100 °C kan bevriezen ter vorming van ijs VII. Sommige vormen van ijs hebben geen eigen
stabiliteitsgebied. Zo is ijs IV een metastabiele vorm die gevormd wordt onder dezelfde
voorwaarden als ijs V. Een kubische vorm van ijs wordt beneden –80 °C direct uit de
dampfase gevormd. Dit is een metastabiele vorm van ijs, alhoewel de dichtheid van kubisch
ijs bijna dezelfde is als de gewone hexagonale vorm van ijs. Kubisch ijs wordt direct in
hexagonaal ijs omgezet bij verwarming boven –80 °C. Kubisch ijs kan zich in zeer hoge
157
wolken vormen. Door hexagonaal ijs bij de temperatuur van vloeibaar stikstof (-196 °C)
samen te drukken, kan een amorfe vorm van ijs worden verkregen.
Figuur 5.4. Fasediagram van water.
5.2.3 Fasediagram van siliciumdioxide
De
polymorfie
van
SiO2
(silica)
is
vrij
complex.
Bij
atmosfeerdruk
en
omgevingstemperaturen is α-kwarts (laag-kwarts) stabiel. Bij 573 °C wordt α-kwarts in βkwarts (hoog-kwarts) omgezet, dat vervolgens op zijn beurt bij 870 °C in tridymiet overgaat.
Boven 1470 °C is cristobaliet de stabiele fase. Cristobaliet smelt bij 1710 °C. De omzetting
van α-kwarts in β-kwarts vindt bij verhitting snel plaats. Dit kan verklaard worden door het
feit dat er voor de omzetting slechts kleine structurele veranderingen noodzakelijk zijn. De
omzetting van β-kwarts naar tridymiet verloopt veel moeizamer, omdat de structurele
veranderingen veel drastischer zijn. Tridymiet is slechts stabiel tot 5 kbar en cristobaliet tot
ca. 8 kbar. De namen laag-kwarts en hoog-kwarts duiden erop dat α-kwarts een lagere
symmetrie (trigonaal) heeft dan β-kwarts (hexagonaal).
Smelten van silica kunnen
gemakkelijk onderkoeld worden ter vorming van kwartsglas. Kwartsglas is heel nuttig voor
het vervaardigen van laboratoriumglaswerk omwille van zijn ongevoeligheid voor
temperatuursveranderingen
en
zuren.
Bovendien
wordt
kwartsglas
gebruikt
voor
ultravioletlampen, cuvetten en optische componenten van spectrofotometers omdat kwartsglas
ultraviolette straling doorlaat. Ook tridymiet en cristobaliet kunnen onderkoeld worden.
Kwartsglas komt in de Natuur voor onder de vorm van lechatelieriet. Dit ontstaat bij
158
blikseminslag op een zandgrond, waarbij de zandkorrels aan elkaar gesmolten worden. Bij
zeer hoge drukken (20 tot 40 kbar) is coesiet stabiel en bij nog hogere drukken (90 tot 120
kbar) komt stishoviet voor. Beide hogedrukpolymorfen komen in de natuur voor om plaatsen
waar zeer hoge drukken en temperaturen zijn opgetreden, zoals bij de impact van meteorieten
op kwartshoudende gesteenten zoals in de Meteor Crater in Arizona (USA). Coesiet (d = 3,00
g cm-3) en stishoviet (d = 4,40 g cm-3) hebben een veel hogere massadichtheid dan α-kwarts
(d = 2,647 g cm-3). Het is opmerkelijk dat coesiet eerst in het laboratorium werd
gesynthetiseerd (in 1953 door Coes) voor het in de Natuur werd gevonden. Het fasediagram
van SiO2 wordt in Figuur 4.5 getoond.
Figuur 5.5. Fasediagram van SiO2.
5.3 Binaire systemen
Bij de systemen met twee componenten (binaire systemen) moet men de concentratie (x)
van de componenten als variabele invoeren. De concentratie wordt in de molaire fracties (of
molfracties) van de componenten uitgedrukt. Voor een binair mengsel met componenten A en
B worden de molfracties xA en xB als volgt gedefinieerd:
159
xA =
aantal mol A
aantal mol A + aantal mol B
(5.4)
xB =
aantal mol B
aantal mol A + aantal mol B
(5.5)
Meestal is A de component met het laagste smeltpunt in het binaire mengsel. In het geval van
mengsels van zuivere metalen drukt men de samenstelling niet uit in molfractie, maar in
atoompercent (at. %). Regelmatig ontmoet men in de literatuur ook fasediagrammen waarbij
op de X-as gewichtspercenten of massapercenten (wt.%) zijn uitgezet. Het massapercent van
component A (WA) en het massapercent van component B (WB) kan men als volgt berekenen:
WA =
massa van component A
× 100
massa van component A + massa van component B
(5.6)
WB =
massa van component B
× 100
massa van component A + massa van component B
(5.7)
Een binaire systeem is alleen volledig weer te geven in een driedimensionele fasefiguur,
met P, T en x als coördinaten. Als de dampspanning laag blijft bij grote temperatuurvariaties
en als de druk niet als variabele hoeft beschouwd te worden, kan de gecondenseerde faseregel
P + F = C + 1 worden gebruikt. Bij binaire systemen (C = 2) komt er onder deze voorwaarden
een nonvariant punt voor (F = 0) wanneer drie fasen met elkaar in evenwicht zijn, een
univariante curve voor twee fasen in evenwicht en een bivariante voorwaarde voor één fase.
De grafische voorstelling van de evenwichtsvoorwaarden in een gecondenseerd systeem met
twee componenten wordt vereenvoudigd door het feit dat er slechts twee variabelen zijn,
namelijk de temperatuur en de samenstelling, en deze kan voorgesteld worden in een
eenvoudig
cartesisch
coördinatensysteem.
Volgens
afspraak
worden
in
binaire
fasediagrammen van gecondenseerde systemen de temperatuur T op de Y-as voorgesteld en
de samenstelling x op de X-as. Dit zijn zogenaamde T-x-diagrammen. De belangrijkste typen
van vast-vloeistof-evenwichten worden hieronder beschreven. We onderscheiden de volgende
drie gevallen:
(1) Alleen de zuivere componenten scheiden zich als vaste stof af.
(2) De beide componenten vormen een verbinding.
(3) De beide componenten vormen mengkristallen.
160
5.3.1 Eenvoudige binaire systemen met een eutecticum
De eenvoudigste binaire fasediagrammen komen voor bij systemen waarbij de
componenten volledig mengbaar zijn in de vloeibare toestand en waarbij alleen de zuivere
componenten zich als vaste stof afscheiden. Als een vloeibaar mengsel van twee
componenten wordt afgekoeld, zal er zich een vaste stof beginnen af scheiden bij een
welbepaalde temperatuur, die men het stolpunt noemt. Bij deze temperatuur zijn twee fasen,
zijnde de vloeistof en vaste stof met elkaar in evenwicht. Volgens de gecondenseerde
faseregel vindt men: F = C - P + 1 = 2- 2 + 1 = 1. Het systeem is dus univariant. Enkel de
temperatuur of de samenstelling van de vloeibare fase is voldoende om het systeem volledig
te definiëren. Als men de stolpunten bepaalt van een reeks van vloeibare mengsels, die in
samenstelling variëren van zuiver A tot zuiver B, en als de stolpunten in een grafiek worden
uitgezet in functie van de samenstelling van de vloeistof, verkrijgt men twee curven die door
AC en BC in Figuur 5.6 zijn weergeven. De punten A en B zijn de stolpunten van de zuivere
componenten A en B. Toevoeging van B aan A verlaagt het stolpunt van de vloeistof volgens
de lijn AC. Toevoeging van A aan B verlaagt het stolpunt van de vloeistof volgens de lijn BC.
Wanneer vloeistoffen rijk aan A worden afgekoeld tot een temperatuur tussen A en C, zal
er zuiver A worden afgescheiden. Voor deze samenstellingen is A de primaire fase, omdat dit
de eerste fase is die bij afkoeling uitkristalliseert. Op eenzelfde wijze zal wanneer vloeistoffen
rijk aan B worden afgekoeld tussen een temperatuur tussen B en C, er zuiver B worden
afgescheiden. Voor deze samenstellingen is B de primaire fase. De curven AC en BC kunnen
dus
beschouwd
worden
als
weergave
van
de
temperatuursvoorwaarden
waarbij
vloeistofmengsels met verschillende samenstellingen in evenwicht staan met resp. vast A en
vast B. Bij punt C, waar de twee curven elkaar raken, staan bijgevolg beide vaste stoffen A en
B in evenwicht met de vloeistof. Aangezien hierbij drie fasen met elkaar in evenwicht staan,
wordt het gecondenseerde systeem nonvariant. Dit is dus de enige temperatuur, bij
atmosfeerdruk, waarbij de vloeibare fase in evenwicht kan staan met de beide vaste stoffen.
Het punt C is de laagste temperatuur waarbij om het even welk vloeibaar mengsel van A en B
zal vast worden, en bijgevolg stelt dit punt het laagste smeltpunt voor van om het even welk
mengsel van vast A en B. Het punt C heet het eutectisch punt (afgeleid van het Grieks:
gemakkelijk smeltend) en het is de laagste temperatuur waarbij de vloeibare fase kan bestaan.
Nu we de betekenis van de lijnen AC en BC, en van het punt C begrijpen, kunnen we de
verschillende gebieden in het fasediagram van Figuur 5.6 nader bekijken. In het gebied boven
de curve ABC kan enkel de vloeibare fase bestaan, omdat de temperatuur gelegen is boven het
smeltpunt van om het even welk mengsel van A en B. De gecondenseerde systemen zijn in dit
gebied bivariant. Er is slechts één fase, de vloeistof, en er zijn twee componenten, zodat er
volgens de gecondenseerde faseregel twee vrijheidsgraden overblijven: F = C - P +1 = 2 – 1 +
161
1 = 2. Het is bijgevolg noodzakelijk om zowel de temperatuur als de samenstelling te
specifiëren om het systeem exact te definiëren. Dit is in overeenstemming met de voorstelling
van zulke systemen door een gebied in het T-x-diagram, waar twee coördinaten vereist zijn
om een systeem te definiëren. Beneden de lijn DCE, waarbij het eutectisch punt werd
gepasseerd, kan er enkel vaste stof voorkomen, aangezien er geen vloeistof beneden de
eutectische temperatuur kan verkregen worden. In dit gebied zijn er twee vaste fasen,
namelijk zuiver A en zuiver B. Volgens de faseregel is het gecondenseerde systeem hier
univariant. De samenstelling van elke fase is vast, zijnde de zuivere component, zodat de
temperatuur de enige variabele is.
Figuur 5.6. Fasediagram van een binair systeem met een eenvoudig eutecticum.
De betekenis van de gebieden ADC en BEC moet nader worden beschouwd. Bij een
bepaalde temperatuur T1 is er een evenwicht tussen de vloeistof met samenstelling x en de
vaste stof voorgesteld door y, die natuurlijk uit zuiver B bestaat. Elk punt tussen x en y stelt
een mengsel voor van vloeistof x en vaste stof y, in evenwicht in verschillende verhoudingen
bij temperatuur T1. Als het punt dichter bij x ligt, dan is er meer vloeistof en minder vaste stof
aanwezig, terwijl als het punt dichter bij y ligt, er relatief meer vaste stof aanwezig is.
Bijvoorbeeld bij z wordt de verhouding van vloeistof tot vaste stof gegeven door de
verhouding van de afstanden yz en xz. Dit is de zogenaamde hefboomregel. Elk punt in het
gebied BEC stelt een evenwicht voor tussen vast B en vloeistof, en elk punt in het gebied
ADC stelt een evenwicht voor tussen vast A en vloeistof. De samenstelling van de fasen in
162
evenwicht kan verkregen worden door een horizontale lijn te trekken door een bepaald punt,
en te bepalen waar deze lijn AD of BE snijdt om de aard van de vaste stof te bepalen, en waar
de lijn AC of BC snijdt om de samenstelling van de vloeistof te bepalen. De lijn xy, en om het
even welke andere horizontale lijn in deze twee gebieden, is een verbindingslijn die de
samenstellingen van de vaste en vloeibare fasen die met elkaar in evenwicht zijn, verbindt. De
curve ACB wordt de stolpuntscurve genoemd., en de curve DCE de smeltpuntscurve. De twee
curven komen samen in het eutectisch punt C, en bijgevolg is bij deze temperatuur de
samenstelling van de vaste stof dezelfde als deze van de vloeistof waaruit deze vaste stof zich
afscheidt. Samenvattend kan men zeggen dat boven de stolpuntscurve, mengsels volledig zijn
opgesmolten, beneden de smeltpuntscurve zijn ze volledig vast, en tussen smeltpuntscurve en
stolpuntscurve treedt gedeeltelijke opsmelting op. In de literatuur wordt de stolpuntscurve ook
wel liquidus genoemd en de smeltpuntscurve solidus.
De term eutecticum wordt niet alleen gebruikt om het nonvariant punt op het fasediagram
aan te duiden, maar ook voor de eutectische samenstelling of eutectisch mengsel. Als men een
eutectisch mengsel opwarmt, gaat het bij de eutectische temperatuur direct in zijn geheel in
de vloeibare fase over, zonder dat er een gedeeltelijke opsmelting plaats vindt. Bij afkoelen
van een gesmolten eutectische samenstelling zal bij overschrijden van de eutectische
temperatuur het mengsel plots als een mengsel van A en B uitkristalliseren, zonder dat er eerst
een primaire fase wordt gevormd. Beneden de eutectische temperatuur kan men geen
vloeibare fase meer waarnemen. Het spreekt voor zich dat de eutectische temperatuur lager is
dan het smeltpunt van zowel zuivere stof A als van zuivere stof B.
Er zijn veel voorbeelden bekend van systemen die twee componenten hebben die volledig
met elkaar mengbaar zijn in de vloeibare fase, maar bij benadering niet mengbaar in de vaste
fase zijn, zodat de twee vaste fasen in zuivere vorm worden uitgescheiden als de vloeistof
wordt afgekoeld. De twee componenten kunnen zijn: metalen, organische verbindingen,
zouten, of een zout en water. Voorbeelden van systemen met een eenvoudig eutectisch punt
zijn Cd ⎯ Zn, Sn ⎯ Zn, Zn ⎯ Al, Ag ⎯ Cu, Sb ⎯ Pb, Si ⎯ Al, Bi ⎯ Cd en AgCl ⎯ KCl.
Toch merkt men vaak op bij metalen dat er een beperkte oplosbaarheid optreedt voor het ene
eindlid in het andere bij hoge temperatuur.
We hebben tot hiertoe steeds aangenomen dat de componenten van een eenvoudig
eutectisch systeem volledig mengbaar zijn in de vloeibare toestand, maar dit is niet altijd het
geval. Het kan zijn dat de vloeistoffen over bepaalde concentratiegebieden niet volledig
mengbaar zijn en dat er twee vloeibare fasen worden gevormd. Zulk een fasediagram wordt in
Figuur 5.7 getoond. Een voorbeeld van een binair systeem dat zich zo gedraagt is zink ⎯
bismut.
163
Figuur 5.7. Fasediagram van een binair systeem met onmengbaarheid in de vloeistoffase.
5.3.2 Binaire systemen met verbindingen
Als de twee componenten A en B van een binair systeem een vaste verbinding AB
vormen, wordt een fasediagram zoals in Figuur 5.8 weergegeven verkregen. Naast de twee
takken AC en BE, die de samenstellingen voorstellen waarbij de vloeistof in evenwicht is met
de vaste stoffen A en B bij verschillende temperaturen, is er een centraal gedeelte CDE dat
een maximum bereikt. Dit gedeelte van het fasediagram stelt vloeistofsystemen voor die in
evenwicht zijn met de vaste verbinding AB. Het maximum van de curve komt steeds voor bij
de samenstelling die overeenkomt met deze van de vaste verbinding, in dit geval met gelijke
hoeveelheden A en B. Het punt D’ is gelegen in het midden tussen zuiver A en zuiver B. Bij
punt D hebben de vloeibare en de vaste fase dezelfde samenstelling, zodat D kan beschouwd
worden als het smeltpunt van de verbinding. Men zegt in dit geval dat verbinding AB een
congruent smeltpunt heeft, aangezien vaste stof en vloeistof met dezelfde samenstelling naast
elkaar kunnen bestaan. Het smeltpunt van AB kan lager, hoger of tussen deze van de twee
zuivere verbindingen A en B gelegen zijn. Het fasediagram in Figuur 5.8 heeft twee
eutectische punten, namelijk C en E. Bij het eutectische punt C scheiden de vaste stoffen A en
AB zich af, terwijl bij eutectisch punt E de vaste stoffen AB en B zich afscheiden. Men kan
een fasediagram met een congruent smeltpunt beschouwen als bestaande uit twee
fasediagrammen van eenvoudige eutectische systemen die naast elkaar worden geplaatst.
Links van de lijn DD’ worden de evenwichtsvoorwaarden gegeven voor het tweecomponentensysteem A-AB, terwijl rechts van DD’ de evenwichtsvoorwaarden voor het
164
twee-componentensysteem AB-B staan weergegeven. Als de twee componenten meer dan één
verbinding vormen, dan zal het fasediagram voor elke verbinding een curve analoog aan CDE
bevatten. In figuur 5.9 wordt het fasediagram getoond voor een binair systeem waarin de
verbindingen A2B
en AB2 met congruente smeltpunten worden gevormd. Hun
samenstellingen en smeltpunten worden door punten D en F gegeven. Dit fasediagram kan
gemakkelijk geïnterpreteerd worden door het te beschouwen als een samenstelling van de drie
afzonderlijke fasediagrammen voor de binaire systemen A⎯ A2B, A2B ⎯ AB2 en AB2 ⎯ B.
Men zou veronderstellen dat als de gevormde verbinding perfect stabiel is bij het smeltpunt en
dus smelt zonder verbinding, de curven een scherp in plaats van een afgerond maximum
vertonen. In bijna alle gevallen treedt er echter een beetje ontbinding van de verbinding in de
samenstellende componenten op. De aanwezigheid van dissociatieproducten in de vloeibare
fase verlaagt het hypothetisch smeltpunt dat de verbinding zou hebben als er geen ontbinding
zou optreden, waardoor de curve wordt afgevlakt. Er bestaan zeer vele voorbeelden van
binaire systemen waarin vaste verbindingen voorkomen. Bijvoorbeeld, aluminium ⎯
magnesium (Al3Mg4), goud ⎯ tin (AuSn) en CaCl2 ⎯ KCl (CaCl2⋅KCl).
Figuur 5.8. Fasediagram van een binair systeem met een congruent smeltende verbinding.
165
Figuur 5.9. Fasediagram van een binair systeem met twee congruent smeltende verbindingen.
Soms is de gevormde verbinding zo onstabiel dat deze reeds volledig ontbindt bij een
temperatuur die beneden zijn smeltpunt is gelegen, zodat de vaste stof niet in evenwicht kan
staan met een vloeibare fase met dezelfde samenstelling. M.a.w., de vaste stof heeft geen echt
smeltpunt. Een fasediagram van dit type wordt in Figuur 5.10 getoond, waarbij er
verondersteld wordt dat er een 1:2 verbinding AB2 wordt gevormd. Bij een temperatuur E
beneden het hypothetisch smeltpunt D, dissocieert de verbinding AB2 volledig in zijn
samenstellende bestanddelen. Volgens CE wordt de vaste stof AB2 afgescheiden, maar
volgens EB vormt B de vaste fase. Het punt E is het incongruente smeltpunt van verbinding
AB2. Als een vloeistof, zoals deze door l wordt voorgesteld, wordt afgekoeld, zal de eerste
vaste stof die zich afscheidt B zijn. Wanneer vervolgens de temperatuur punt E bereikt, begint
de verbinding AB2 zich te vormen. Aangezien er nu één vloeistof en twee vaste fasen zijn, is
het system nonvariant, en de temperatuur bij E zal constant blijven totdat alle vaste stof B
volledig in de vaste verbinding is omgezet of totdat alle vloeistof is verdwenen. Voor alle
punten rechts van D, zal dit laatste gebeuren, omdat dan het originele systeem rijker is aan B
dan aan de verbinding AB2 en er onvoldoende A aanwezig is om alle vaste stof B in AB2 om
te zetten. Wanneer de samenstelling van het systeem tussen D en E is gelegen, zal de vaste
stof die zich heeft afgezet volledig in AB2 worden omgezet, en daarna zal de temperatuur
verder dalen tot het eutectische punt C wordt bereikt. Punt E wordt vaak een transitiepunt
genoemd. Voorbeelden van systemen waarin een incongruent smeltende verbinding
166
voorkomt, zijn Na ⎯ K (Na2K), natrium ⎯ bismut (NaBi), goud ⎯ antimoon (AuSb2) en
KCl ⎯ CuCl2 (2KCl⋅CuCl2).
Figuur 5.10. Fasediagram van een binair systeem met een incongruent smeltende verbinding.
5.3.3 Binaire systemen met vaste oplossingen
Het eenvoudigste vaste-oplossingssysteem is datgene waarbij de beide componenten in de
vaste fase in elke verhouding mengbaar zijn (Figuur 5.11). Er komt slechts een vaste fase en
slechts één vloeibare fase voor. Beide componenten vormen bijgevolg een ononderbroken
reeks vaste oplossingen (of mengkristallen). Dit type van fasediagram wordt bijvoorbeeld
gevonden voor de binaire mengsels goud ⎯ zilver, AgCl ⎯ NaCl en PbCl2 ⎯ PbBr2. Het
smeltpunt van het ene eindlid, B, wordt verlaagd door toevoeging van het andere eindlid, A,
en omgekeerd, zodat het smeltpunt van A toeneemt door toevoeging van B. De stolpuntscurve
en smeltpuntscurve zijn gelijkmatige lijnen die elkaar enkel bij de samenstelling van de
eindleden A en B raken. Er komt geen discontinuïteit in het fasediagram voor. Het geheel van
stolpuntscurve en smeltpuntscurve noemt men ook smeltlus. Bij lage temperaturen komt er
een één-fase vaste oplossing voor en het systeem is bivariant (C = 2, P = 1, en bijgevolg F =
2). Bij hogere temperaturen komt er een één-fase vloeibare oplossing voor, die eveneens
bivariant is. Bij intermediaire temperaturen komt er een twee-fasengebied van een vaste
oplossing + vloeistof voor. De samenstelling van de vaste fase verandert continu met die van
de vloeibare fase waaruit de fase fase wordt afgescheiden. Bij een gegeven temperatuur is de
vloeistof relatief rijker dan de vaste fase aan die component wiens toevoeging het stolpunt
167
verlaagt. Als een vloeistof met samenstelling l wordt afgekoeld, begint deze te stollen bij punt
y, en de samenstelling van de vaste fase die zich vormt wordt door z gegeven. In dit geval
verlaagt toevoeging van A het stolpunt, zodat de vloeistof rijker is aan A dan de vaste fase.
Aangezien de vaste fase rijker is aan B dan de vloeistof, wordt de vloeistof bij afkoeling rijker
aan A en de temperatuur volgt de hele tijd de stolpuntscurve yA. De toestand van het systeem
als geheel volgt de lijn ls. De samenstellingen van de vloeistof en van de vaste oplossingen in
evenwicht met de vloeistof bij een bepaalde temperatuur, kunnen gevonden worden door het
trekken van een horizontale verbindingslijn door dat punt. Het snijpunt met de stolpuntscurve
L geeft de samenstelling van de vloeistof en het snijpunt met de smeltpuntscurve S geeft de
samenstelling van de vaste fase. Naarmate de temperatuur daalt, stijgt ook de hoeveelheid
vaste stof in vergelijking met de hoeveelheid vloeistof. Als het systeem punt z’ bereikt staan
de laatste resten vloeistof met samenstelling y’ op het punt te verdwijnen. Bij deze
temperatuur is het stolproces juist voltooid. Merk echter op dat dit resultaat enkel wordt
verkregen als de vloeibare en vaste fase steeds met elkaar in evenwicht blijven. Dit is vaak
niet het geval, want kristallen van de vaste oplossing kunnen aan het systeem worden
onttrokken.
Figuur 5.11. Continue reeks van vaste oplossingen
168
Bij een systeem met vaste oplossing is scheiding van de zuivere componenten door
gefractioneerde kristallisatie mogelijk, juist zoals een mengsel van vloeistoffen door
gefractioneerde kristallisatie kan gescheiden worden (Figuur 5.12). Een vloeistof met
samenstelling l begint te kristalliseren ter vorming van een vaste oplossing bij punt a”. Als
het mengsel verder wordt afgekoeld tot punt x, bestaat het mengsel voor ongeveer gelijke
delen uit vloeistof a en vaste stof b. De vloeistof is rijker aan A en de vaste stof is rijker aan B
dan het oorspronkelijke mengsel. Als de laag a nu wordt afgescheiden en verder tot x’
afgekoeld, verkrijgt men gelijke hoeveelheden van vloeistof a’ en vaste stof b’. Door het
proces te herhalen kan men op den duur zuiver A verkrijgen. Als de vaste stof b wordt
afgescheiden, opgesmolten en opnieuw tot punt x” afgekoeld, zal het systeem bestaan uit
gelijke hoeveelheden a” en b”. Door deze procedure met de vaste stof te herhalen kan na een
paar kristallisaties zuiver B worden verkregen.
Figuur 5.12. Gefractioneerde kristallisatie van vaste oplossingen.
In binaire systemen met een vaste oplossing, worden vaak metastabiele of nietevenwichtsproducten gevormd. Dit fenomeen treedt telkens op als de afkoeling van de smelt
zo snel verloopt dat het evenwicht zich niet telkens bij elke temperatuur opnieuw kan
instellen. De kristallen die zich eerst vormen bij afkoeling van een vloeistof b hebben de
samenstelling a. Als de kristallen van de vaste oplossing niet de tijd hebben om terug in
evenwicht te komen met de vloeistof, worden ze bij verdere afkoeling definitief aan het
systeem onttrokken. Elk nieuw kristal dat neerslaat wordt een beetje rijker aan B en het
169
resultaat is dat er zich kristallen vormen met een samenstelling die varieert van a tot ergens
tussen b en B. Vaak hebben de kristallen die neerslaan tijdens het afkoelingsproces een
gelaagde structuur. Het centrale gedeelte dat zich eerst vormt heeft een samenstelling a en de
opeenvolgende afzetting worden geleidelijk rijker aan B als men verder van de kristalkern
weg gaat.
De vorming van kristallen met een gelaagde structuur neemt men vaak waar bij
mineralen. De plagioklaasveldspaten zijn vaste oplossingen van anorthiet, CaAl2Si2O8 en
albiet, NaAlSi3O8. Het overeenkomstige fasediagram wordt in Figuur 5.13 getoond. Vele
stollingsgesteenten bevatten grote kristallen van plagioklazen als ze bij de vorming traag
werden afgekoeld. Plagioklaassmelten en –kristallen bereiken echter slechts zeer traag de
evenwichtsvoorwaarden. Alhoewel stollingsgesteenten in de natuur vaak de kans kregen om
zeer traag af te koelen, is het niet uitzonderlijk om gesteenten aan te treffen waarbij
gefractioneerde kristallisatie heeft plaats gevonden. De plagioklaaskristallen die hierbij
ontstonden hebben een calciumrijke kern en natriumrijke buitenste zones.
Figuur 5.13. Fasediagram van het systeem anorthiet-albiet.
Sommige typen van eenvoudig fasediagram van vaste oplossingen vertonen een thermisch
maximum of minimum in de stolpuntscurve en smeltpuntscurve (Figuur 5.14). Deze maxima
en minima worden indifferentiële punten genoemd omdat ze geen echte nonvariante punten
170
zijn. Bij een nonvariant punt staan drie fasen met elkaar in evenwicht (F = 0, P = C + 1 = 3).
Deze voorwaarde wordt echt nooit bereikt voor systemen met een vaste oplossing omdat er
nooit meer dan twee fasen aanwezig zijn, namelijk een vaste oplossing en een vloeibare
oplossing. De stolpuntscurve en smeltpuntscurve verlopen continu door het maximum of
minimum en vertonen geen discontinuïteit (in tegenstelling met peritectica en eutectica).
Dit type van fasediagrammen heeft eenzelfde uitzicht als deze van vloeistofmengsels die
azeotropen vormen. Daarom noemt men een minimum ook wel eens een azeotropisch
minimum en een maximum een azeotropisch maximum. Bij verhitting van mengkristallen met
de azeotropische samenstelling zal een scherp smeltpunt optreden. Alhoewel een binair
fasediagram met een minimum lijkt op een binair fasediagram met een eenvoudig eutecticum,
is er toch een belangrijk verschil. Als men een vloeistof met de azeotropische samenstelling
laat uitkristalliseren, verkrijgt men een homogeen mengkristal. Als men echter een vloeistof
met de eutectische samenstelling laat uitkristalliseren, is de gestolde massa echter nooit
homogeen maar bestaat uit een mengsel van kristallen van zuiver A en kristallen van zuiver
B. Minima komen in fasediagrammen van mengkristallen veel voor. Voorbeelden zijn Cu ⎯
Mn, Cu ⎯ Au, Mn ⎯ Ni, As ⎯ Sb, KCl ⎯ KBr, Na2CO3 ⎯ K2CO3, HgBr2 ⎯ HgJ2.
Maxima zijn daarentegen zeer zeldzaam; men kent ze enkel bij mengsels van enkele d- en lvormen van organische stoffen. Het maximum ligt dan juist bij de samenstelling van het
racemische mengsel.
Figuur 5.14. Fasediagrammen van binaire vaste oplossingen met een thermisch minimum
(links) en een thermisch maximium (rechts).
Het gebeurt vaak dat twee verbindingen slechts over een beperkte samenstellingsgebied
een vaste oplossing met elkaar kunnen vormen, zodat er tussen twee concentratiegrenzen
naargelang de temperatuur twee geconjugeerde vaste oplossingen kunnen bestaan. In vaste
stof A kan een zekere hoeveelheid van vaste stof B oplossen ter vorming van een bepaalde
vaste oplossing, terwijl er in vaste stof B een kleine hoeveelheid van vaste stof A kan
171
oplossen ter vorming van een andere vaste oplossing. Buiten bepaalde concentratiegrenzen
worden er twee vaste fasen gevormd, die elk een homogene oplossing van A en B zijn.
Aangezien het nu mogelijk is om drie fase te hebben, twee vaste fasen en een vloeistof, zullen
de curven die het vast-vloeistof-evenwicht weergeven een discontinuïteit vertonen.
Naargelang de situatie kan een eutectisch punt of een transitiepunt optreden.
Een fasediagram met een eutectisch punt wordt in Figuur 5.15 getoond. AC en BC zijn de
stolpuntscurven, terwijl AD en BE de smeltpuntscurven zijn. Volgens AD kunnen de vaste
stoffen beschouwd worden als vaste oplossingen van B met een overmaat A (S1-type) en
volgens BE bestaan de vaste oplossingen grotendeels uit B met een gedeelte A (S2-type). De
stolpuntscurven raken elkaar in C, dat het eutectische punt van het systeem is. Het verschilt
op één vlak van het eutectisch punt in een eenvoudig binair eutectisch systeem: de vaste fasen
die in evenwicht staan met de vloeistof zijn geen zuivere verbindingen, maar twee vaste
oplossingen die door punten D en E worden voorgesteld. Het eutectisch punt is echter een
nonvariant punt in het systeem en het is de laagste temperatuur waarbij vloeistof kan bestaan.
Het gebied FDEG is het ontmenggebied van de vaste oplossingen. Fasediagrammen van dit
type zijn intermediair tussen deze waarbij de verbindingen volledig onmengbaar zijn in de
vaste toestand en deze die een continue reeks van vaste oplossingen met een
smeltpuntsminimum vormen. Als de vaste stoffen in Figuur 5.15 beter en beter met elkaar
mengbaar worden, zullen de punten D en E uiteindelijk met elkaar samenvallen en verkrijgen
we het fasediagram van een vaste oplossing met smeltpuntsminimum. Als anderzijds de vaste
stoffen minder en minder mengbaar worden, wordt de scheiding tussen D en E groter totdat
de samenstellingen met deze van zuiver A en B samenvallen en men het fasediagram van een
eenvoudig eutectisch systeem verkrijgt. Juist zoals de oplosbaarheden van gedeeltelijk
mengbare vloeistoffen temperatuursafhankelijk is, zal ook de wederzijdse oplosbaarheid van
vaste stoffen A en B temperatuursafhankelijk zijn. In het algemeen zal de wederzijdse
oplosbaarheid van de vaste stoffen dalen naarmate de temperatuur daalt, en de voorwaarden
voor de vast-vast-evenwichten worden door de twee lijnen DF en EG weergegeven. De
samenstelling van de twee vaste oplossingen die met elkaar in evenwicht zijn bij een bepaalde
temperatuur kan gevonden worden door een horizontale verbindingslijn te tekenen van DF
naar EF bij die welbepaalde temperatuur. Bijvoorbeeld, de vaste stoffen p en q zijn met elkaar
in evenwicht. Fasediagrammen zoals weergegeven in Figuur 5.15 worden gevonden voor de
systemen goud ⎯ nikkel, bismut ⎯ lood, cadmium ⎯ tin, ijzer ⎯ chroom, KNO3 ⎯ TlNO3
en AgCl ⎯ CuCl.
Een fasediagram met een transitiepunt (peritectisch punt) wordt in Figuur 5.16
getoond. In dit geval leidt toevoeging van B tot een stijging van het smeltpunt van A, zoals
men kan verwachten voor vaste oplossingen, maar er zijn echter wel goed gedefinieerde
172
mengbaarheidslimieten. De stolpuntscurve AC geeft de samenstellingen van vloeistoffasen die
in evenwicht zijn met vaste oplossingen van het S1-type volgens de smeltpuntcurve AD, en op
eenzelfde wijze is BC de stolpuntscurve die overeenkomt met de smeltpuntcurve BE voor
vaste oplossingen van het S2-type. Het punt C stelt een nonvariant gecondenseerd systeem
voor, aangezien hier drie fasen in evenwicht zijn, zijnde twee vast oplossingen en een
vloeistof. Dit punt wordt een transitiepunt of een peritectisch punt genoemd. Terwijl een
eutectisch punt steeds gelegen is tussen de samenstellingslimieten van de vaste fasen die in
evenwicht zijn met de vloeistof, ligt de samenstelling van een eutectisch punt steeds buiten
deze samenstellingslimieten. Bij een peritectisch punt vormt het snijpunt van de
stolpuntscurven geen minimum, dit in tegenstelling tot het geval van een eutectisch punt.
Binaire systemen die twee reeksen van vaste oplossingen met een transitiepunt vertonen zijn
cadmium ⎯ kwik, AgCl ⎯ LiCl en AgNO3 ⎯ NaNO3
Figuur 5.15. Fasediagram van gedeeltelijk mengbare vaste fasen met een eutectisch punt.
173
Figuur 5.16. Fasediagram van gedeeltelijk mengbare vaste stoffen met een peritectisch punt.
5.3.4 Binaire systemen met vast-vast fasetransities
Eerste-ordefasetransities in de vaste toestand die een verandering van een eigenschap
zoals het volume of enthalpie inhouden, kunnen op eenzelfde manier worden behandeld als
smeltprocessen. In een één-component gecondenseerd systeem, bijv. zuiver A, kunnen twee
polymorfen met elkaar in evenwicht bestaan bij één vast punt. In een binair systeem zonder
vaste oplossingen, kunnen fasetransities in ieder van de twee eindleden A en B door
horizontale lijnen (isothermen) worden weergegeven. In Figuur 5.17 worden de laagtemperatuurpolymorfen van A en B aangeduid met αA en αB. De transitietemperaturen zijn
T1 voor de transitie αA ' βA, T2 voor transitie αA ' βA en T4 voor transitie βA ' δA. De
transitietemperaturen voor de fasemengsels zijn dezelfde als deze voor de zuivere fasen.
174
Figuur 5.17. Fasediagram van een binair systeem met vast-vast fasetransities.
5.4 Ternaire systemen
Ternaire systemen bestaan uit drie componenten. Voor ternaire systemen zijn er vier
vrijheidsgraden, aangezien de temperatuur, druk en de samenstelling van twee componenten
onafhankelijke veranderlijken kunnen zijn. De aanwezigheid van een derde component vereist
in vergelijking met de binaire systemen een aanpassing van de methode voor de grafische
weergave van zowel temperatuur als samenstelling. De drie componenten worden weergeven
op de hoekpunten van een driehoekig coördinatensysteem. Elk hoekpunt stelt 100% van één
van de componenten voor. Beschouw een systeem X-Y-Z (Figuur 5.18). Een binaire
samenstelling zoals A die weergegeven wordt op een zijde van de driehoek tussen X en Y,
stelt een mengsel van X en Y voor. Aangezien het punt A halfweg gelegen is tussen X en Y,
komt het overeen met 50 % X en 50 % Y. De samenstelling van punt B komt overeen met
ongeveer 80 % Z en 20 % X.
175
Figuur 5.18. Samenstellingsdriehoek waarbij de methode wordt getoond om de samenstelling
van punt C te bepalen in het ternaire systeem X-Y-Z.
Ternaire samenstellingen zoals C kunnen bepaald worden door vanuit dit punt loodlijnen
te trekken naar elk van de zijden (Figuur 5.18). De lengte van elke loodlijn is evenredig met
het percentage van de component die weergegeven wordt door het hoekpunt tegenover de
zijde die door de loodlijn wordt gesneden. Punt C stelt een samenstelling voor bestaande uit
10 % X, 60 % Y en 30 % Z. Een eenvoudigere methode om de samenstelling van een ternair
mengsel af te leiden wordt in Figuur 5.19 getoond. Doorheen punt C worden twee lijnen
getekend die elk parallel zijn met één van twee zijden van de driehoek. Deze twee lijnen
snijden de derde zijde, waarbij deze zijde in drie delen wordt verdeeld. De relatieve lengte
van de drie delen correspondeert met de relatieve hoeveelheden van de drie componenten
weergegeven door punt C. De eenvoudigste methode is om het ternaire fasediagram te
tekenen op speciaal grafiekpapier. In dit geval kan de samenstelling gemakkelijk worden
afgelezen, zoals getoond in Figuur 5.20.
176
Figuur 5.19. Bepaling van de samenstelling van punt C door de tweelijnenmethode. Doorheen
C worden twee lijnen getekend die parallel zijn met twee vrij te kiezen zijden van de driehoek
(in dit geval XY en YZ). De snijpunten van deze twee lijnen met de derde zijde (XZ) verdeelt
deze zijde in drie delen waarvan de lengten evenredig zijn met de relatieve hoeveelheden van
de componenten X, Y en Z in punt C.
Figuur 5.20. Het punt C op driehoekig grafiekpapier. Het percentage van de componenten X,
Y en Z kan direct afgelezen worden van de genummerde coördinaatsassen.
De temperatuurcoördinaat staat loodrecht op het driehoekig samenstellingsrooster. De
meeste ternaire systemen die men beschouwt zijn gecondenseerde systemen, zodat de
dampspanning en dus de druk kan verwaarloosd worden. Wanneer de stolpuntsvlakken
worden getekend, wordt een driedimensionele figuur gecreëerd (Figuur 5.21). Alhoewel
177
driedimensionele modellen van ternaire fasediagrammen kunnen worden getekend of
geconstrueerd, is zulk een model niet gemakkelijk te interpreteren. De gebruikelijke
procedure is om de samenstellingen in een driehoek weer te geven en om de vorm van de
verschillende stolpuntsvlakken te tonen door middel van lijnen van gelijke temperatuur
(isothermen) zoals op een topografische kaart. Bovendien worden de grenzen tussen de
stolpuntsvlakken van de verschillende vaste fasen afgelijnd door middel van dikke lijnen die
men de grenslijnen noemt. Zulk een representatie is te zien in het fasediagram van het ternaire
systeem CaSiO3 – CaSiTiO5 – CaAl2SiO8 (Figuur 5.22). Aangezien er in dit systeem geen
vaste oplossingen voorkomen, is dit fasediagram de combinatie van drie eenvoudige binaire
systemen. De isothermen nemen in temperatuur toe in de richting van de smeltpunten van de
drie componenten. De dikke lijnen die de grenzen tussen de stolpuntsvlakken aflijnen zijn
temperatuursminima in het systeem. De grenslijnen verdelen het diagram in gebieden die de
primaire gebieden worden genoemd en die in feite de stolpuntsvlakken zijn van de
componenten in het systeem. De grenslijnen kunnen gezien worden als een uitbreiding van de
binaire eutectica naar een ternair systeem. De drie grenslijnen snijden elkaar in een
temperatuursminimum dat een ternair eutecticum wordt genoemd. Om het ternair
fasediagram te vereenvoudigen worden vaak de isothermen niet getoond. Ternaire
fasediagrammen kunnen behoorlijk ingewikkeld worden als er vaste oplossingen en
intermediaire verbindingen aanwezig zijn. Het ternair systeem CaO – Al2O3 – SiO2 is een
voorbeeld van een vrij complex systeem (Figuur 5.23).
Figuur 5.21. Temperatuur-samenstellingsmodel van een ternair systeem X-Y-Z. Elk van de
drie verticale zijden is een binair systeem. De bovenkant toont de stolpuntsvlakken.
178
Figuur 5.22. Fasediagram van het ternair systeem CaSiO3 – CaSiTiO5 – CaAl2SiO8 met
temperatuurscontouren om de stolpuntsvlakken aan te duiden. De drie primaire fasegebieden
worden gescheiden door de grenslijnen (dikke lijnen) die elkaar snijden op een punt met een
minimale temperatuur (ternair eutecticum).
Figuur 5.23. Fasediagram van het ternaire systeem CaO – Al2O3 – SiO2,
waarop de primaire gebieden worden getoond.
179
Hoofdstuk 6:
Synthesemethoden in de vaste-stofchemie
De methoden voor de bereiding van materialen in de vaste toestand zijn vrij verschillend
van de synthesemethoden van discrete moleculen. De eigenschappen van anorganische en
organische moleculaire systemen kunnen gewijzigd worden door reactie in oplossing, waarbij
bepaalde groepen worden verwijderd en toegevoegd en waarbij de producten bijvoorbeeld
door kristallisatie of via chromatografische methoden kunnen worden gezuiverd. Bij de
synthese van vaste stoffen gaat het over de behandeling van het gehele rooster. Vaak is een
zuivering van de verbindingen na de synthese niet meer mogelijk omwille van de lage
oplosbaarheid van de gevormde fasen. Daarom moet er goed op letten om een overmaat aan
reagentia te vermijden.
6.1 Hoge-temperatuursmethoden (keramische methoden)
De meest gebruikte methode voor de bereiding van vaste stoffen is door reactie van de
vaste componenten in de juiste molaire verhoudingen op hoge temperatuur gedurende een
lange periode. Vele gemengde metaaloxiden, metaalchalcogeniden (sulfiden, seleniden en
telluriden) en metaalpnictiden (nitriden en fosfiden) werden op deze manier gesynthetiseerd.
Zeer nauwkeurig afwegen van de startproducten is van primordiaal belang omdat de
gevormde producten niet na synthese kunnen worden gezuiverd. Daarom zijn hygroscopische
stoffen (waterabsorberend) en niet-stoechiometrische stoffen (variabele samenstelling) bij de
keuze van de startproducten te vermijden. Indien mogelijk moet men ook vluchtige en
luchtgevoelige startproducten vermijden. Als er geen alternatief is, moeten afgesloten buizen
worden gebruikt.
Aangezien vaste-stofreacties worden uitgevoerd bij temperaturen tot 2000 °C, is klassiek
laboratoriumglaswerk niet bruikbaar. Bijvoorbeeld, borosilicaatglaswerk (Pyrex®) wordt
reeds bij 600 °C week. Daarom worden materialen met een hoog smeltpunt gebruikt, zoals
capsules uit edelmetalen (goud, platina), kroesjes uit alumina en zirconia, en buizen of
kroesjes uit kwartsglas gebruikt. Deze keuze van het materiaal voor de reactiebehouder hangt
af van het type van reagentia en van de temperatuur.
De klassieke verwarmingsmethoden die in het syntheselabo worden gebruikt zoals een
verwarmingsmantel of een oliebad, zijn niet in staat om de hoge temperaturen te leveren
waarbij vaste stoffen met elkaar reageren. Zeer hoge temperaturen (tot 4000 °C) kunnen met
behulp van een laser worden bereikt, maar gelukkig zijn voor de meeste vaste-stofreacties
zulke extreme temperaturen niet vereist. Temperaturen tot 2000 °C kunnen worden bereikt in
oven die een elektrische weerstandsdraad gebruiken om de vereiste temperatuur te bereiken.
180
Een moffeloven verhit een vrij groot volume tot op een vastgestelde hoge temperatuur. Een
buisoven heeft een kleiner verhit gebied in het midden van de buis (typisch 2-5 cm) en de
temperatuur daalt snel naar de uiteinden van de buis toe. Een buisoven kan ook gebruikt
worden om stalen te verhitten in een reactieve gasatmosfeer door de buisoven te verbinden
met een glasfles.
De keramische synthesemethoden zijn zeer traag. Dit komt vooral omdat er geen smelt
wordt gevormd en de gehele reactie in de vaste fase moet verlopen. Dit vereist diffusie van de
atomen langsheen de contactpunten tussen de vaste deeltjes. Het reactiemengsel bestaat uit
een verzameling van kleine deeltjes en tussen de deeltjes bevinden zich open ruimten. De
kationen migreren naar de contactpunten en vormen de nieuwe structuur op het grensvlak
(Figuur 6.1). Meestal zijn de kationen veel mobieler dan de anionen, omdat ze veel kleiner
dan de anionen zijn. Naarmate er meer nieuw product op het grensvlak wordt gevormd,
moeten de kationen verder migreren doorheen het gevormde product om de nieuwe fase te
vormen. De reactie wordt trager en trager naarmate het diffusiepad langer en langer wordt.
Figuur 6.1. Schematische voorstelling van een vaste-stofreactie waarbij de reactie door
diffusie van ionen op het contactoppervlak plaatsvindt.
De reactie kan versneld worden door de lengte van het diffusiepad korter te maken en
door het aantal contactpunten tussen de deeltjes in met mengsel te doen toenemen. Dit kan op
een aantal manieren verwezenlijkt worden. Zo kan men het reactiemengsel regelmatig
hermalen om de homogeniteit te verbeteren. Het contact tussen de deeltjes van de reagentie
kan verbeterd worden door de deeltjes dichter bij elkaar te dwingen, bijvoorbeeld door het
mengsel tot een pil samen te persen om het aantal holten tussen de deeltjes te verminderen.
181
Door verhoging van de temperaturen neemt de migratiesnelheid van de kationen toe. In de
praktijk wordt een combinatie van deze drie methoden gebruikt.
De poeders die als startproducten voor de keramische methode worden gebruikt moeten
zo fijn mogelijk worden gemalen om de specifieke oppervlakte te vergroten. De vastestofreactie vindt immers alleen maar aan het contactoppervlak plaats. De specifieke
oppervlakte is sterk afhankelijk van de deeltjesgrootte. Dit kan door een eenvoudige
berekening worden aangetoond. Een kubisch kristal met een volume van 1 cm3 heeft 6 zijden
met een oppervlakte van 1 cm2 en bijgevolg is de totale oppervlakte 6 cm2. Wanneer dit
kristal nu 10× parallel aan ieder vlak wordt gesneden, verkrijgt men 103 kubische kristallieten
met elk een dimensie van 0,1 × 0,1 × 0,1 cm. Deze 103 kleine kubusjes hebben dezelfde massa
en volume als de grote kubus, maar hun totale oppervlakte is 10× groter (103 × 6 × 0.01 cm3).
Wanneer men een kristal met een volume van 1 cm3 gedurende een zekere tijd in een
kogelmolen maalt, verkrijgen de deeltjes een gemiddelde grootte van ongeveer 10 μm (10-3
cm). Bijgevolg is de totale oppervlakte van het poeder 6 × 103 cm2 (0,6 m2) als alle deeltjes
kubische kristallieten zijn (Figuur 6.2). Merk op dat bij een deeltjesgrootte van 10 μm de
diffusieafstanden bij reactie nog meer dan 104 eenheidscellen bedragen. Het is duidelijk dat
verkleining van de deeltjesgrootte tot nanometerafmetingen grote voordelen kan bieden naar
de reactiesnelheden van vaste-stofsynthese toe.
Figuur 6.2. Wanneer een kubisch kristal met een ribbelengte van 1 cm (totale oppervlakte 6
cm2) 10× parallel aan iedere zijde wordt gesneden, verkrijgt men 103 kubische kristallieten
met een ribbelengte van 0,1 cm. Het totale volume blijft hetzelfde, maar de totale oppervlakte
neemt toe tot 60 cm2.
De lange reactietijd is maar één van de problemen die met de keramische methode
verbonden zijn. Het vinden van de juiste reactietemperatuur is meestal niet eenvoudig en moet
voor nieuwe verbindingen door trial-and-error worden bepaald. Vaak is het gevormde
product niet volledig homogeen, zelfs niet wanneer de reactie bijna volledig opgaat. Als er op
182
het einde van de reactie een onzuiverheid achterblijft, is deze bijna onmogelijk om te
verwijderen. Zo kan bij een vaste-stofreactie tussen BaCO3 en TiO2 naast het gewenste
BaTiO3 (bariumtitanaat) ook BaTi2O5 als nevenproduct worden gevormd. Het is heel moeilijk
om een homogene verdeling te verkrijgen van de doterende stoffen die zo belangrijk zijn in
vaste stoffen zoals anorganische fosforen. In vele systemen kan de reactietemperatuur niet zo
hoog worden gebracht als gewenst voor een redelijke reactiesnelheid, omdat anders één of
meer componenten uit het reactiemengsel zouden kunnen vervluchtigen.
Als voorbeeld van een vaste-stofsynthese kan de bereiding van de hoge-temperatuur
keramische supergeleider YBa2Cu3O7-x worden beschouwd. Deze verbinding kan gevormd
worden door herhitting van een mengsel van yttriumoxide (Y2O3), bariumoxide (BaO) en
koper(II)oxide (CuO). De startproducten moet fijn gemalen en goed gemengd worden om het
specifiek oppervlak en de contactgebieden tussen tussen de korrels te vergroten. Het
poedervormig mengsel wordt tot pillen geperst om een goed contact tussen de korrels te
krijgen. De geperste pillen worden in aan schuitje uit alumina geplaatst en in een oven
langszaam over een periode van 8 tot 12 uren tot 930 °C opgewarmd. Het reactiemengsel
wordt gedurende 12 tot 16 uren bij 930 °C gehouden, vervolgens tot 500 °C afgekoeld en
gedurende 12 tot 16 uren op een constante temperatuur van 500 °C gehouden. Daarna laat
men het mengsel tot kamertemperatuur afkoelen. Na verhitting op 930 °C is de samenstelling
van het staal ongeveer YBa2Cu3O6,5. Door het product vervolgens een tijd bij 500 °C te
houden, reageert het verder met zuurstof uit de lucht en de uiteindelijke samenstelling is
ongeveer YBa2Cu3O6,9. Het zuurstofgehalte x in YBa2Cu3O7-x is sterk afhankelijk van de
partiële zuurstofdruk tijdens deze verwarmingsstap bij 500 °C. De laatste stap mag niet bij te
hoge temperatuur worden uitgevoerd (350-500 °C), omdat anders de opname van zuurstof
veel moeilijker verloopt. De basisreactie is die de vorming van het stoechiometrische
YBa2Cu3O7 beschrijft is:
C , 500° C
Y2O3 + 4BaO + 6CuO + ½ O2 ⎯950
⎯°⎯
⎯
⎯→ 2YBa2Cu3O7
Het is ook mogelijk om andere precursoren te gebruiken voor de synthese van YBa2Cu3O7-x.
Zo kan Ba(NO3)2 gebruikt worden als een bron van barium. Dit omzeilt het probleem dat
BaO koolstofdioxide uit de lucht absorbeert ter vorming van BaCO3 dat moeilijk te ontbinden
is.
De in-situ ontbinding van Ba(NO3)2 geeft fijncorrelig BaO met een groot specifiek
oppervlak en een verhoogde reactiviteit. In plaats van nitraten, wordt bij vaste-stofreacties
ook gebruik gemaakt van carbonaten en andere oxyzouten die op hoge temperatuur
ontbinden. Het temperatuursprogramma waaraan het reactiemengsel wordt onderworpen is
sterk afhankelijk van de vorm en de reactiviteit van de reagentia. Als één of meerdere
183
reagentia een oxozout is, wordt het mengsel eerst voor een paar uren tot een geschikte
temperatuur verhit om de ontbinding van de reagentia op een gecontroleerde manier te laten
verlopen. Als deze stap achterwege wordt gelaten en het mengsel direct tot op hoge
temperatuur wordt verhit, kan de thermische ontbinding van de reagentia erg heftig verlopen.
YBa2Cu3O7-x is een supergeleider bij ongeveer –181 °C, een temperatuur die duidelijk boven
de temperatuur van vloeibare stikstof (-196 °C) is gelegen. De eenheidscel van YBa2Cu3O7-x
wordt in Figuur 6.3 getoond. Deze structuur kan van de perovskietstructuur afgeleid worden
(zie hoofdstuk 3). De kristalstructuur van YBa2Cu3O7-x is een elektrondeficiënte variatie van
de perovskietstructuur.
Figuur 6.3. Schematische voorstelling van de vorming van de structuur van YBa2Cu3O7-x
uitgaande van deze van spinel.
Eén van de grootste problemen met vaste stofreacties is dat de reagentia niet op atomaire
schaal gemengd zijn. In plaats van een mechanisch gemengd poedervormig mengsel te
gebruiken, kan men het startmengsel via de coprecipitatiemethode bereiden. Hierbij worden
de verschillende metaalkationen samen uit een oplossing neergeslagen. De neerslag bestaat
dan uit een zeer innig mengsel van de zouten of uit een vaste oplossing. De zouten van de
verschillende metalen worden in eenzelfde solvent (meestal water) opgelost. Vervolgens
worden de metalen neergeslagen door de oplossing in te dampen of door toevoeging van een
neerslagvormend reagens. Bij deze methode worden vaak hydroxiden, carbonaten, oxalaten,
formiaten of citraten gevormd. De verkregen neerslag wordt verhit bij de geschikte
temperatuur en in een geschikte atmosfeer om het eindproduct te verkrijgen. De
184
coprecipitatiemethode is enkel succesvol als de metaalzouten een gelijkaardige oplosbaarheid
hebben en als de neerslagsnelheid gelijkmatig is of vaste oplossingen worden gevormd. Deze
methode kan aan de hand van de synthese van ZnFe2O4 spinel worden geïllustreerd. Hierbij
worden zink(II)oxalaat en ijzer(III)oxalaat afzonderlijk in de verhouding 1:1 in water
opgelost. De twee oplossingen worden dan gemengd en verhit om het water te verdampen. De
neergeslagen fijne poeder is een vaste oplossing die de kationen op atomaire schaal vermengd
bevat. Het poeder wordt afgefiltreerd en verhit. Omwille van de hoge graad van
homogenisatie,
zijn
in
vergelijking
met
de
keramische
methode
veel
lagere
reactietemperaturen vereist om de reactie te laten verlopen. De algehele reactie kan
geschreven worden als:
Fe2(C2O4)3 + Zn(C2O4) → ZnFe2O4 + 4CO + 4CO2.
Een andere voorbeeld is de vorming van de gemengde oxiden M1-xM’xO (M, M’ = Ca, Mg,
Mn, Fe, Co, Zn, Cd). Omdat de carbonaten van deze metalen isostructureel zijn
(calcietstructuur), kunnen een groot aantal carbonaathoudende vaste oplossingen met als
formule M1-xM’x(CO3) die twee (of meer) kationen bevatten, worden bereid. Deze vaste
oplossingen zijn ideale precursoren voor de synthese van vaste oplossingen van de
overeenkomstige oxiden M1-xM’xO met de NaCl-structuur. De carbonaten worden thermisch
ontbonden in vacuüm of in een stroom van droog stikstofgas. De eenvoudige vorming van de
oxiden uitgaande van de carbonaten is te wijten aan de structurele overeenkomsten tussen de
calciet- en de NaCl-structuur. De M1-xM’xO vaste oplossingen kunnen gebruikt worden als
precursoren voor de synthese van spinellen of andere complexe oxiden. Een groot aantal
ternaire en kwaternaire metaaloxiden kan bereid worden uitgaande van vaste oplossingen met
hydroxide-, nitraat- of cyanide-ionen. Zo kunnen vaste oplossingen met de algemene formule
Ln1-xMx(OH)3 (Ln = La, of Nd: M = Al, Cr, Fe, Co of Ni) en La1-x-yNixMy(OH)3 (M = Co of
Cu) in de Ln(OH)3-structuur worden gekristalliseerd. De verbindingen kunnen op relatief lage
temperaturen (rond 600 °C) worden ontbonden ter vorming van LnNiO3, LaNi1-xCoxO3,
LaNi1-xCuxO3, .... Een variatie op de coprecipitatiemethode is de precursormethode waarbij
stoechiometrische gemengde metaalzouten worden neergeslagen. Hierbij zijn de kationen in
de juiste verhouding eenzelfde vaste precursor ingebouwd. Bijvoorbeeld, ferrietspinellen
zoals NiFe2O4 kunnen bereid worden door trage verhitting van het gemengde metaalacetaat
Ni3Fe6O3(OH)(OAc)17⋅2py (OAc = acetaat, py = pyridine) tot 200-300°C om het organisch
materiaal weg te branden, gevolgd door verwarming in lucht op ongeveer 1000°C gedurende
2 tot 3 dagen. Het gemengde zout dat als startproduct wordt gebruikt heeft een Ni:Fe
verhouding van exact 1:2 zoals in het gewenste eindproduct. Een ander type van gemengde
185
metaalprecursoren zijn redoxverbindingen. Verbindingen zoals (NH4)2Cr2O7 die zowel het
oxiderende Cr2O72--ion als het reducerende NH4+-ion bevatten, ontbinden autokatalytisch tot
Cr2O3 wanneer ze in brand worden gestoken (“kunstmatige vulkaan”). Het exotherm karakter
van de reactie is te wijten aan de oxidatie van het ammoniumion tot N2 en H2O door het
dichromaat-ion dat zelf tot chroom(III) wordt gereduceerd:
(NH4)2Cr2O7 → N2 + 4H2O + Cr2O3
Bij deze reactie worden fijne volumineuze gasdeeltjes verkregen door de gasvormende
reactie. Gelijkaardige precursoren werden gebruikt voor de synthese van chromietspinellen,
MCr2O4 (M = Mg, Zn, Cu, Mn, Fe, Co, Ni). Bijvoorbeeld, magnesiumchromiet, MgCr2O4,
wordt gesynthetiseerd door neergeslagen (NH4)2Mg(CrO4)2⋅6H2O geleidelijk tot 1100-1200
°C te verhitten:
(NH4)2Mg(CrO4)2⋅6H2O → MgCr2O4 + N2 + 10 H2O
Door
een
nauwkeurige
controle
van
de
experimentele
voorwaarden
zijn
deze
precursormethoden in staat om fasen met een zeer accurate stoechiometrie te geven. Dit is
belangrijk omdat verscheidene chromieten en ferrieten waardevolle magnetische materialen
zijn waarvan de eigenschappen gevoelig zijn voor de zuiverheid en de stoechiometrie.
6.2 Verbrandingssynthese
De verbrandingssynthese maakt gebruikt van zeer exotherme reacties. Zulke reacties
hebben typisch een zeer grote activeringsenergie en zetten grote hoeveelheden energie vrij.
Eens de reactie geïnitieerd is door de de snelle energie-input van een uitwendige bron, komt
er voldoende energie vrij om de reacties zelfstandig te laten onderhouden. De reagentia
worden snel verhit (103 tot 106 K/s) tot zeer hoge temperaturen. De reacties zijn zo snel dat
alle energie die wordt geproduceerd door de exotherme reactie wordt gebruikt om het staal te
verhitten. Het doen ontbranden van de reactie kan gebeuren met een laserpuls, met een
elektrische boog, een elektrisch vonk, door een chemische reactie, ... In de zelf-propagerende
variant van de verbrandingssynthese (de “zelfpropagerende synthese op hoge temperatuur”)
De verbrandingsreactie wordt op een welbepaald punt in het reactimengsel geïnitieerd en
verplaatst zich vervolgens snel door het reactiemengsel in de vorm van een verbrandingsgolf.
In de gelijktijdige verbrandingsvariant, die ook “thermische explosie” wordt genoemd, wordt
186
het gehele staal eerst tot de verbrandingstemperatuur verhit, waarna de reactie gelijktijdig in
het gehele staal plaatsvindt eerder dan dat er een verbrandingsgolf optreedt.
De verbandingssynthese biedt een aantal voordelen ten opzichte van de conventionele
keramische methode. Er is minder energie vereist dan bij de conventionele methoden. De
korte reactietijden zorgen voor lage verwerkingskosten en tijdsbesparing. Er is geen dure
apparatuur vereist. Anorganische materialen kunnen in een stap door gebruik te maken van de
chemische energie vrijgezet door de reagerende reagentia gesynthetiseerd en in de
uiteindelijke vorm gebracht worden. Door de hoge reactietemperaturen worden vluchtige
onzuiverheden uitgedreven, wat resulteert in producten met een hogere zuiverheid. De grote
thermische gradiënten en de snelle afkoelsnelheden kunnen aanleiding geven tot de vorming
van thermodynamisch niet-stabiele fasen. Dit is een verschil met de keramische
synthesemethoden waarbij door de lange reactietijden bijna steeds de thermodynamisch
stabiele fasen worden gevormd.
Verbrandingssyntheses zijn moeilijk onder controle te houden door de grote
reactiesnelheden. Een zekere controle is mogelijk door toevoeging van verdunningsmiddelen
die niet aan de reactie deelnemen, maar die de thermische massa van het systeem doet
toenemen. Het verdunningsmiddel kan het product zijn (bijv. de snelheid van de zeer
exotherme reactie van titaan en boor kan verminderd worden door toevoeging van TiB2) of
kan een inerte verbinding zijn. In het laatste geval wordt er een composietmateriaal gevormd.
Een andere mogelijkheid om de reactie onder controle te houden is door de korrelgrootte van
de reagentia te vergroten. De leidt tot een lage snelheid van de verbrandingsgolf, waardoor de
reactiesnelheid daalt.
De materialen die worden verkregen met behulp van verbrandingssynthese variëren van
brosse, licht gesinterde poeders tot monolitische poreuze producten die worden gevormd als
slechts een fractie van de producten is gesmolten bij de piektemperatuur. De inherente grote
porositeit van de producten vindt zijn oorsprong in het verschil in massadichtheid tussen de
reagentia en de producten en door het uitdrijven van opgeloste gassen of verdamping door de
hoge reactietemperaturen. Belangrijke parameters die een invloed hebben op de samenstelling
en morfologie van het einddproduct zijn: (a) de deeltjesgrootteverdeling, morfologie en
zuiverheid van de startproducten; (b) de massadichtheid van het reactiemengsel, die een
invloed heeft op de thermische geleidbaarheid van het mengsel; (c) de ontstekingstechniek, de
begintemperatuur en de druk van het reactiemengsel; (d) het temperatuursverschil tussen het
initiële reactiemengsel en de volledig gereageerde producten; (e) de grootte van het staal en
de configuratie van het reactievat. Verdichting van het eindproduct kan verkregen worden
door het samen te persen als het nog in de plastische toestand is op de hoge
verbrandingstemperaturen.
187
Een vroege toepassing van verbrandingssynthese is het thermietlassen om spoorstaven
aan elkaar te verbinden. Hiervoor wordt gebruik gemaakt van een mengsel van ijzer(II)oxide
en aluminium (= thermiet). Als het wordt aangestoken treedt er een chemische reactie op
waarbij zeer veel warmte vrijkomt. De temperatuur kan tijdens de reactie tot 3000 °C stijgen.
Hierbij ontstaat metallisch ijzer en aluminiumoxide:
Fe2O3 + 2Al → 2Fe + Al2O3
De reactie verloopt vlot maar niet explosief. Daar thermiet pas bij ca. 1500 C tot ontbranding
komt, moet de reactie door een ontstekingspatroon worden ingeleid. Deze bestaat uit
aluminiumpoeder, gemengd met een oxidatiemiddel als kaliumchloraat of bariumperoxide.
Het thermietmengsel kan ook door middel van een brandend magnesiumlint tot ontbranding
worden gebracht. Thermiet brandt zelfs onder water nog door. De warmte die door deze
exotherme reactie wordt gegeneerd (ΔH = -851,5 kJ/mol) is voldoende om spoorstaven aan
elkaar te lassen. Eenmaal aangestoken kan de reactie niet geblust worden - in deze reactie is
er sprake van een overdracht van een of meerdere zuurstof-atomen, dus er wordt geen
zuurstof uit de lucht gebruikt. Met water blussen heeft vaak zelfs een omgekeerd effect: door
de hoge temperatuur dissocieert water in zuurstof en waterstof, waardoor de reactie alleen
maar heviger wordt en er zelfs explosiegevaar ontstaat. Bij het thermietlassen wordt een mal
uit vuurvast materiaal om de twee aan elkaar te lassen spoorstaven bevestigd. Binnen deze
mal vermengt het residu van het thermiet, dat hoofdzakelijk uit vloeibaar ijzer bestaat, zich bij
een hoge temperatuur met het ijzer van de spoorstaven. Voor het verkrijgen van een goede
verbinding is het van belang dat de beide spoorstaven goed zijn uitgelijnd, de mal goed sluit
en de verbinding voldoende is afgekoeld voordat deze belast wordt. Een andere term voor
thermietlassen is aluminothermie. Thermiet kan gebruikt worden voor het lassen onder water.
Thermiet wordt ook in brandbommen toegepast. De ontdekking van het thermietmengsel
werd gemaakt in 1893 door de chemicus Hans Goldschmidt.
Alhoewel het eigenlijke thermiet een mengsel is uit ijzer(II)oxide en aluminium, wordt de
term ook gebruikt voor andere mengsels van aluminium met metaaloxiden (bijv. CuO, TiO2,
Sb2O3, ...). In plaats van aluminiumpoeder kan ook magnesiumpoeder worden gebruikt.
Magnesium is zelfs beter, omdat het gevormde MgO-bijproduct gemakkelijk met een
waterstofchloride-oplossing kan worden opgelost. Naast de klassieke thermietreactie waarbij
een metaaloxide tot het metaal wordt gereduceerd, is het ook mogelijk om de reductie van een
oxide tot een element te laten volgen door de reactie met een ander element ter vorming van
moeilijke smeltbare verbindingen zoals boriden, carbiden, siliciden en nitriden. Voorbeelden
zijn:
188
SiO2 + C + 2Mg → SiC + 2MgO
TiO2 + B2O3 + 5Mg → TiB2 + 5MgO
Vele carbiden, siliciden, boriden, nitriden, oxiden of hydriden kunnen door middel van
verbrandingssynthese uit de samenstellende elementen worden verkregen. Zo is de reactie
tussen titaan en koolstof ter vorming van titaancarbide één van de best bestudeerde
verbrandingssynthesen. De reactievergelijking is:
Ti + C → TiC
De synthese kan in 2kg batchen worden uitgevoerd. Titaancarbide is een nuttig vuurvast
materiaal en slijppoeder.
Een techniek die verwant is met verbrandingssynthese is de metathesereactie in de vaste
fase (solid state metathesis, SSM). Hierbij worden de reagentia zodanig gekozen dat één van
de producten (gewoonlijk een bijproduct) een zeer grote roosterenergie heeft, waardoor de
vorming van dit product de drijvende kracht voor de reactie is. Een voorbeeld is:
5Li3N + 3TaCl5 → 3TaN + 15LiCl + N2
Covalent tantaal(V)chloride reageert met reactief lithiumnitride ter vorming van twee stabiele
producten, tantaalnitride en lithiumchloride. Lithiumchloride kan gemakkelijk worden
opgelost waardoor zuiver tantaalnitride achterblijft.
6.3 Hydrothermale synthese
Bij een hydrothermale synthesemethode worden de reagentia in water of stoom bij hoge
temperaturen en drukken verhit. Het water heeft twee functies, namelijk drukdoorgevend
midden en als een solvent, waarin de oplosbaarheid van de reagentia druk- en
temperatuursafhankelijk is. De methode is heel eenvoudig. De reagentia en water worden
geplaatst in een dikwandige metalen cilinder, die men “bom” of autoclaaf noemt, die bestand
is tegen hoge drukken en temperaturen en die hermetisch kan worden afgesloten. Om
aantasting van het metaal van de autoclaafwand te vermijden, is de binnenwand vaak bekleed
met teflon, zilver, goud of platina. De autoclaaf wordt vervolgens in een oven geplaatst,
189
gewoonlijk bij een temperatuur tussen 100 en 700 °C. Boven 374 °C gaat water over naar een
superkritische toestand, waarbij er geen verschil meer is tussen de vloeibare en gasvormige
toestand. De drukken in een autoclaaf kunnen oplopen van 200 tot 3000 atmosfeer. De druk in
de autoclaaf kan geregeld worden door het vulpercentage van het reactievat: hoe meer het vat
met vloeistof is gevuld, hoe groter de uiteindelijke druk zal zijn (Figuur 6.4). De inhoud van
de autoclaaf wordt gedurende zekere tijd op een constante hoge temperatuur gehouden of
wordt gecontroleerd traag afgekoeld. Men kan de autoclaaf ook aan een temperatuursgradiënt
onderwerpen, waardoor de bovenkant van de autoclaaf bij een lagere temperatuur wordt
gehouden dan de onderkant.
Figuur 6.4. Druk in een autoclaaf als functie van de temperatuur en de vulpercentage.
Een groot aantal klassen van verbindingen kunnen met behulp van de hydrothermale
synthese worden gesynthetiseerd, waaronder oxiden, hydroxiden, wolframaten, molybdaten,
carbonaten en silicaten. Typische verbindingen die via deze methode worden gesynthetiseerd
zijn zeolieten en synthetisch kwarts. De methode is vooral geschikt om verbindingen te
synthetiseren die in water bij kamertemperatuur slechts in heel beperkte mate oplosbaar zijn.
Bij verhoogde temperatuur en druk, en dan vooral in de superkritische toestand heeft water
een groter oplossend vermogen voor anorganische verbindingen. Als de oplosbaarheid van de
uit te kristalliseren stof bij verhoogde temperatuur nog te laag is, kan aan de oplossing een
zogenaamde mineraliseerder toegevoegd worden. Zo verhoogt toevoeging van NaOH aan
water de oplosbaarheid van SiO2 in water zeer sterk. De methode wordt gebruikt voor de
synthese van verbindingen die niet stabiel zijn bij hun smeltpunt (incongruent smeltende
190
verbindingen) of verbindingen met een hoge dampdruk bij hun smeltpunt. Hydrothermale
synthese is uitermate geschikt voor de groei van grote kristallen met een goede kwaliteit,
zoals synthetische kwartskristallen die voor piëzo-elektrische toepassingen. gebruikt worden
191
Bronvermelding
De cursustekst werd samengesteld op basis van de volgende boeken:
Atkins, P.; Overton, T.; Rourke, J.; Weller, M.; Armstrong, F., Shriver & Atkins Inorganic
Chemistry, 4th Edition (Oxford University Press, 2006).
Barrett, J., Inorganic Chemistry in Aqueous Solution, Tutorial Chemistry Texts Vol. 21
(Royal Society of Chemistry, Cambridge, UK, 2003).
Bloss, F.D., Crystallography and Crystal Chemistry: an Introduction (Holt, New York,
1971).
Büchler, K.H.; Moretto, H.H.; Woditsch, P., Industrial Inorganic Chemistry, 2nd Edition
(Wiley-VCH, Weinheim, 2000).
Dann, S.E., Reactions and Characterization of Solids, Tutorial Chemistry Texts Vol. 2 (Royal
Society of Chemistry, Cambridge, UK, 2000).
Ehlers, E.G., The Interpretation of Geological Phase Diagrams (Freeman, San Francisco,
1972).
Glasstone, S., Textbook of Physical Chemistry, 2nd Edition (MacMillan, London, 1968).
Greenwood, N.N.; Earnshaw, A., Chemistry of the Elements, 2nd Edition (ButterworthHeinemann, Oxford, 1997).
Harris, D.C., Quantitative Chemical Analysis, 5th Edition (Freeman, New York, 1999).
Heller, G.; Vervest, J.C.G.; Wilbrink, H.F., Vademecum van de Glastechniek (Kluwer,
Deventer, 1992).
Housecroft, C. E.; Sharpe, A. G. Inorganic Chemistry (Pearson, Harlow, 2001).
Huheey, J. F.; Keiter, E.A.; Keiter, R.L., Inorganic Chemistry, Principles of Structure and
Reactivity, 4th Edition (HarperColins College Publishers, New York, 1993)
192
Hurlbut, C.S. jr.; Klein, C., Manual of Mineralogy, 19th Edition (Wiley, New York, 1977).
Miessler, G. L.; Tarr, D. A. Inorganic Chemistry, 2nd Edition (Prentice Hall, Upper Saddle
River, 1999).
Moffatt, W.G.; Pearsall, G.W.; Wulff, J., Materialen 1: Structuur (Reeks Prisma-Technica
deel 25) (Het Spectrum, Utrecht, 1969).
Mortimer, C.E., Chemistry, 6th Edition (Wadsworth Publishing Company,1986).
Müller, U., Inorganic Structural Chemistry, 2nd Edition (Wiley, Chichester, 2007).
Ramdohr, P.; Strunz, H., Klockmanns Lehrbuch der Mineralogie, 16. Auflage (Ferdinand
Enke Verlag, Stuttgart, 1978).
Shriver, D.F.; Atkins, P.W., Langford, C.H.
Inorganic Chemistry, 1st Edition (Oxford
University Press, 1990).
Shriver, D.F.; Atkins, P.W., Inorganic Chemistry, 3rd Edition (Oxford University Press,
1999).
Schubert, U.; Hüsing, N., Synthesis of Inorganic Materials (Wiley-VCH, Weinheim, 2000).
Verbrugh, A.J., Elementaire Inleiding tot de Fysische Scheikunde, Deel 1 (Wolters,
Groningen, 1962).
West, A. R., Basic Solid State Chemistry, 2nd Edition (Wiley, Chichester, 1999).
Wiberg, N., Holleman-Wiberg Lehrbuch der Anorganischen Chemie, 91th Edition (Walter de
Gruyter, Berlin, 1985).
193